سندرم کریگلر نجار (CNS) نوعی اختلال نادر و خطرناک است که به صورت ارثی بوده و به کبد مربوط میشود. اسم این سندرم برگرفته از اسم یابندگان آن در سال ۱۹۵۲ به نامهای جان کریگر و ویکتور نجار است. آنچه در این بیماری اتفاق میافتد، افزایش سطح نوعی سم به نام بیلیروبین در خون است که «هایپربیلیروبینمی» نام دارد.
بیلیروبین حاصل شکستن گلبولهای قرمز خون در یک چرخه نرمال است. به منظور حذف آن از بدن، این ماده به کبد میرود. در کبد آنزیمی به نام اوریدین فسفات گلوکورونوزیل ترانسفراز (UGT) روی آن اثر میکند و طی واکنشی از نوع کنژوگاسیون آن را از نوع سمی بیلیروبین به مادهای قابل دفع تبدیل میشود. این ماده سپس میتواند از طریق صفرا و روده از بدن خارج میشود.
در اختلال CNS، آنزیم UGT در بدن یا به کل تولید نمیشود (نوع I) و یا به شدت کاهش مییابد (نوعII). نتیجه هر رو نوع اختلال عدم شکستن کامل بیلیروبین است که مانع دفع آن به داخل صفرا میشود.
تجمع غیر عادی بیلیروبین غیر کنژوگه در خون منجر به یرقان میشود و در نتیجه ممکن است به سمت مغز مهاجرت کرده و اختلالهای شدید مغزی مانند کرنایکتروس را سبب میشود.
CNS نوع I که در آن مقدار تولیدی آنزیم UGT در بدن تقریبا نزدیک صفر است، بیماری بوجود آمده شدیدتر میباشد و اگر در سنین پایینتر رخ دهد، بسیار کشنده است. در سندرم کریگلر نجار نوع II که بدن بیمار قادربه تولید سطح پایینی از UGT میباشد، بیماری خفیفتر بوده و معمولا منجر به کرنایکتروس نمیشود و به دارو پاسخ میدهد.
به عکسهای زیر توجه کنید:

سطح غیر طبیعی سطح طبیعی
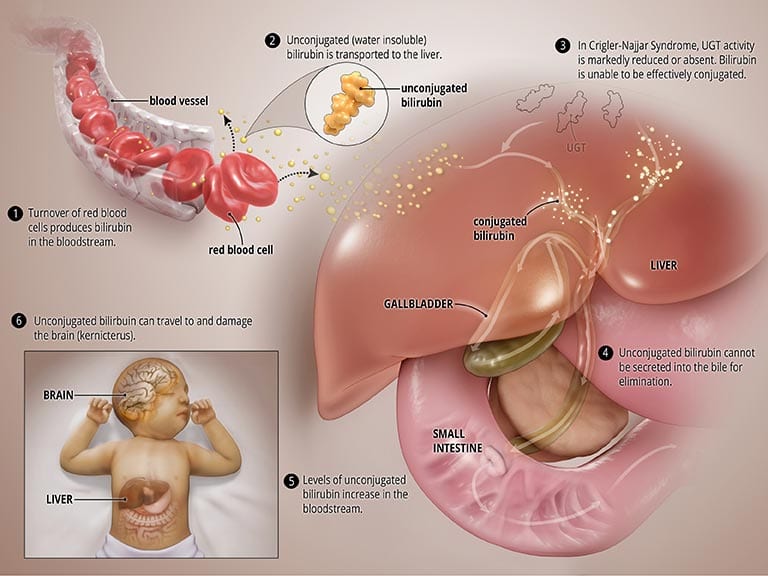

علت سندرم کریگلر نجار چیست؟
CNS نتیجه ایجاد جهش در ژن تولید کننده آنزیم UGT در کبد ، یعنی ژن UGT1A1 است. نوع وراثت این ژن به صورت اتوزومال مغلوب میباشد، به این معنی که کودک باید از هر دو والدین کروموزوم معیوب را دریافت کند تا علائم بیماری بروز کند. اگر این اتفاق بیفتد و کودک به سندرم کریگلر نجار مبتلا شود، انزیم UGT تولید نمیشود (نوعI) و یا به شدت کاهش مییابد (نوعII) که باعث تجمع بیلیروبین در بدن میشود.
اگر ژن معیوب تنها از یک والد به کودک منتقل شود و دیگر کروموزوم کودک دارای ژن سالم باشد، نوع خفیفی از بیماری به نام سندرم ژیلبرت برای کودک اتفاق خواهد افتاد.
آیا این سندرم رایج میباشد؟
تخمینها نشان میدهند که تعداد کودکان مبتلا به سندرم کریگلر نجار در کل دنیا، کمتر از ۱ کودک بین هر یک میلیون کودک متولد شده است! به علت زمینه ژنی و نوع وراثت این نشانگان، برای ابتلای کودک به CNS لازم است که هر دو والدین حداقل یک ژن جهش یافته داشته باشند.
علائم سندرم کریگلر نجار
علائم نشانگان کریگلر نجار نوع I تقریبا اندکی پس از تولد خود را نشان میدهند. کودک مبتلا دچار یرقان شدید و زردی پوست، غشاهای مخاطی و سفیده چشم میشود. این علائم تا بعد از سه هفته اول پس از تولد نیز ادامه مییابد.
در ماه اول زندگی نوزاد ، احتمال ابتلا به کرنایکتروس یا همان انسفالوپاتی بیلیروبین بالا میرود.
کرنایکتروس یک بیماری خطرناک نورولوژیکی میباشد که بیلیروبین سمی در مغز تجمع یافته و بیشتر میشود و به سیستم عصبی مرکزی آسیب وارد میکند. از علائم مربوط به کرنایکتروس میتوان به کاهش سطح انرژی (لتارژی یا بیحالی)، استفراغ، تب و احتمالا تغذیه نامطلوب اشاره کرد.
دیگر علائم این بیماری به نبود برخی از رفلکسها در بدن نوزاد مربوط میشوند (رفلکس مورو)؛ اسپاسمهای ملایم تا شدید عضلات: مانند شرایطی که در آن سر و پاشنهها به عقب خم شده و بدن به سمت جلو تحدب پیدا میکند (اپیستوتونوس) ، به همراه حرکات غیر قابل کنترل و غیر ارادی عضلات (اسپاستیکس).
به علاوه، حرکات مک زدن و شیر خوردن نوزاد ممکن است کاهش یابد، صدای گریه او شدیدتر و بلندتر میشود و تون عضلات او ممکن است کاهش یابد (هیپوتونی) که در نتیجه آن شلی غیرعادی در کودک دیده میشود.
علائم بیماری کرنایکتروس میتواند خفیفتر نیز باشد، علائمی مانند خامدستی، مشکلات مهارتهای ظریف حرکتی و عدم تکامل کامل مینای دندان. علائم شدیدتر آن شامل از دست رفتن شنوایی، اختلال در درک حسی، تشنج و حرکات نامنظم ، آهسته ، مداوم و غیر ارادی (آتتوز یا حرکت پریشی) دستها و پاها و کل بدن.
اختلال کرنایکتروس درنتیجه آسیب وخیم مغزی حاصل میشود.
با این که اختلال کرنایکتروس معمولا در نخستین دوران کودکی خود را نشان میدهد ولی در برخی موارد مربوط به CNS نوع I این ناهنجاری ممکن است تا اواخر دوران کودکی یا اوایل بزرگسالی بروز نکند.
بیمارانی که در برههای از زندگیشان جهت تثبیت سطح بیلیروبین خون در محدوده نرمال تحت فوتوتراپی قرار میگیرند، در صورت قطع درمان و یا قرار گرفتن در معرض دیگر بیماریها ممکن است به اختلال کرنایکتروس دچار شوند.
سندرم کریگلر نجار نوع II خفیفتر از نوع I میباشد. در برخی از موارد درگیر نیز تا زمان بزرگسالی تشخیص داده نمیشود. کودکان مبتلا دچار یرقان میشوند که با گذشت زمان و در صورت بیماری به صورت همزمان، در صورت نخوردن غذا به مدت طولانی و یا قرار گرفتن تحت بیهوشی ممکن است شدیدتر شود.
معمولا سندرم کریگلر نجار نوع II به ندرت منجر به کرنایکتروس میشود ولی در صورت داشتن بیماری، گشنه ماندن و داشتن بیهوشی ریسک آن افزایش مییابد.
چگونگی تشخیص CNS
موارد شدید یرقان نوزادی معمولا مشکوک به کرنایکتروس میباشند. تایید دقیق آن وابسته به ارزیابیهای کلینیکی، تاریخچه خانوادگی و تستهای آزمایشگاهی و ژنتیکی است. به طور مثال، پزشک با درخواست تست خون و مشاهده مقدار بالای بیلیروبین غیر کنژوگه خون و یا کمبود بیلیروبین کنژوگه در صفرا میتواند تشخیص دقیقتری ارائه دهد.
تست ژنتیک نیز برای بررسی جهشها و موتاسیونهای رخ داده در ژن UGT1A1 لازم است.
درمان سندرم کریگلر نجار
هدف اولیه راههای درمانی طی شده در CNS، کاهش سریع و دائمی میزان بیلیروبین غیرکنژوگه خون میباشد. این پروسهها بسته به اینکه CNS نوع I میباشد یا نوع II به روش های مختلفی طی میشوند.
در CNS نوع I درمان خط اول فوتوتراپی میباشد. در این روش کودک در یک دستگاه مشابه تخت برنزه خوابیده و در معرض تابش نور LED آبی قرار میگیرد. نور تابیده شده راهی میانبر برای نیاز بدن به کنژوگاسیون به شمار میآید؛ بدین ترتیب که بیلیروبین غیر کنژوگه را شکسته و امکان ورود آن به صفرا و دفع از راه روده را فراهم میکند.
با این حال فوتوتراپی پروسهای خسته کننده است. این روش روزانه ۱۰ الی ۱۲ ساعت طول میکشد. علاوه بر این، به علت اینکه تابش طولانی نور به بدن باعث ضخیم شدن پوست میشود، افزایش شدت و قدرت فوتوتراپی را به مرور زمان طلب میکند. نیاز به فوتوتراپی به شکل چشمگیری کیفیت زندگی را کاهش میدهد.
پیوند کبد یکی از مؤثرترین راههای درمانی برای بیماران مبتلا به سندرم کریگلر نجار میباشد. یک کبد جدید آنزیمهای لازم برای تبدیل بیلیروبین غیر کنژوگه به بیلیروبین کنژوگه را خواهد داشت ( یعنی تغییر فرم غیرقابل دفع به فرم قابل دفع از بدن).
با این حال بیماران همچنان ژن معیوب و جهش یافتهای که موجب کمبود گلوکورونیل ترنسفراز میشود را دارند و میتوانند طی تولید مثل به نسل بعدی منتقل کنند.
برخی موارد مبتلا به CNS نوع II نیز ممکن است نیاز باشد در مراحل شدید هایپربیلیروبینمی با فوتوتراپی درمان شوند. برخی نیز با مصرف روزانه فنوباربیتال موفق به کنترل این ناهنجاری شدهاند.
پیش آگهی سندرم کریگلر نجار
با در پی گرفتن راههای درمانی مناسب، بیماران مبتلا به CNS نوع II میتوانند تقریبا زندگی نرمالی داشته باشند.
متاسفانه بیماران مبتلا به CNS نوع I با مشکلات بیشتری روبهرو میشوند و احتمال آسیبهای مغزی شدید و غیرقابل بازگشت در این بیماران بیشتر است. تنها راه درمانی موجود برای این نوع از اختلال تنها در برهه خاصی از کودکی مؤثر میباشد. در نتیجه نیاز به پیوند و درمانهای جایگزین برای این بیماری بسیار ضروری به نظر میرسد.
آینده امیدوار کننده برای درمان CNS
مطالعات انجام گرفته در مورد راههای درمانی سندرم کریگلر نجار و جبران کردن کمبود آنزیمهای مربوطه، در حال پیشرفت است. آزمایشهای کلینیکال ژن درمانی که در آن تلاش میشود ژن معیوب UGT1A1 را با نسخه سالم آن جایگزین کنند تا بدن بتواند عملکرد نرمالش را بدست بیاورد، در حال مطالعه است. اگر ژن درمانی در موارد کلینی نیز جواب دهد، میتواند درمانی دائمی و همیشگی برای این بیماران باشد.
یکی دیگر از حوضههای درمانی مربوط به پیوند سلولهای کبدی نرمال به بیماران مبتلا به نشانگان کریگلر نجار برای تولید مقدار کافی آنزیم برای جبران کمبود ژن سالم UGT1A1 میباشد. با این حال، پیوند سلولی کبد سالم نیاز همیشگی به سرکوب سیستم ایمنی خواهد داشت: مشابه روند پیوند کبد.