ژنوم سرطان
شناسایی جامع ژنوم تومورها به محض توالی یابی ژنوم انسانی در سال ۲۰۰۱ به هدف اصلی پژوهشگران تبدیل شد. از آن زمان به بعد، پیشرفت تکنولوژی توالی و ابزارهای تحلیلی باعث شده است تا این زمینه تحقیقاتی شکوفا شود.
در شش مقاله ۶-۱ در این شماره از Nature، تجزیه و تحلیل پان سرطان کل ژنوم ها (PCAWG)جامع ترین و کامل ترین متا آنالیز ژنوم های سرطان را در کنسرسیوم ارائه می دهد. تلاش های قبلی که عمدتاً روی مناطق کد کننده پروتئین ژنوم سرطان متمرکز بود؛ PCAWG ژنوم کامل را تجزیه و تحلیل می کند. هر مقاله جنبه مهمی از ژنتیک سرطان را بررسی می کند – در کنار هم، یافته های آن ها برای درک پیچیدگی ژنتیکی کامل سرطان مهم است.
پروژه ی نقشه ی ژنوم سرطان
قبل ازتفسیر تأثیر این تحلیل ها، بولد کردن دیتاها و چارچوب سازمانی پیچیده ای که زیربنای تلاشPCAWG می باشد، بسیارمهم است. این پروژه شامل یک گروه بین رشته ای از دانشمندان ۴ قاره، متشکل از ۷۴۴ نفر، که برای محافظت از داده های بیمار می بایست با چالش های عمده فنی، حقوقی و اخلاقی برای انجام تجزیه و تحلیل های توزیع شده مواجهه شوند. محققان به ۱۶ گروه کاری تقسیم شدند ؛که هر یک روی جنبه های مشخص ژنومیک سرطان متمرکز بشدند – برای مثال ارزیابی پیک جهش ها، یا به نحوه ی تکامل تومور پرداختند.
در مجموع کنسرسیوم تجزیه و تحلیل های یکپارچه از ۳۸ نوع تومور انجام شده است. این گروه، توالی ۲۶۵۸ ژنوم سرطانی (PCAWG)، در کنار نمونه های همسان سلول های غیر سرطانی از همان افراد را دارا می باشند. این داده ها توسط ۱۱۸۸ ترانسکریپتور که توالی یابی آر ان آ های سرطانی را انجام می دادند، تکمیل شده است.
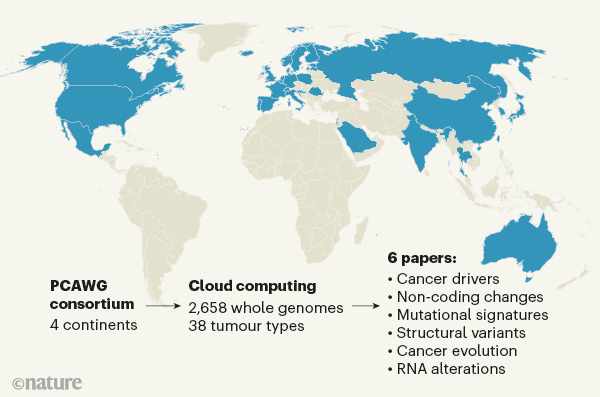
این تلاش ها شامل کنترل کیفیت گسترده، پردازش داده های هماهنگ، همچنین اعتبار سنجی تجربی گسترده و منظم pipeline محاسباتی مورد استفاده برای شناسایی جهش ها بود. بسیاری از الگوریتم های محاسباتی و Pipeline درتوالی مورد استفاده قرار گرفتند. این امر به صدها ترابایت داده نیاز داشت، در چندین مرکز داده و احتمالاً طی میلیون ها ساعت پردازش – که همه از طریق محاسبات ابری – تسهیل می شوند. به ویژه تلاش های PCAWG مثال مهمی از چگونگی محاسبات ابری است که همکاری بین المللی را ممکن ساخته و به پیشرفت آن کمک کند.
مقاله مرتبط: سرطان چیست و سرطان چگونه باعث مرگ می شود؟
مقالات پروژه ی ژنوم سرطان
۱-اولین مقاله موجود نمای کلی گستردگی مجموعه داده ها را ارائه می دهد. کنسرسیوم گزارش می دهد که به طور متوسط در هر ژنوم سرطانی، چهار یا پنج جهش (driver mutation) رخ می دهد، که به سلول های سرطانی یک مزیت انتخابی می دهد. فقط 5٪ تومورهای مورد مطالعه این جهش را نشان ندادند در مقابل، بسیاری از سرطان ها نشانههایی از کاتاستروفی ژنومی به نام های کروموپلکسی (۱۷.۸٪ تومورها) و کروموتریپسیس (۲۳.۳٪) را به نمایش گذاشتند که منجر به تغییرات عمده ساختاری در ژنوم می شوند.
پنج مقاله دیگر با جزئیات بیشتری به جنبه های مختلف مجموعه داده ها می پردازند. به عنوان مثال:
۲-در مقاله دوم، راینبی و همکاران به شناسایی محرک های ژنتیکی غیر کد کننده ی دی ان آ پرداختند، اگرچه یک اقدام بلند پروازانه بود؛ زیرا تشخیص دقیق جهش ها در مناطق کد نویسی نشده نسبت به مناطق کدشده، یا ارزیابی آن ها بسیار مشکل است. نویسندگان از مدل سازی دقیق برای حذف آثار قدیمی و شناسایی سیستماتیک جهش ها(non-coding driver mutations) غیر کد کننده استفاده کردند.
نتایج پیشین توالی های غیر کدگذاری شده ی RNA های طولانی بدون کد نویسی NEAT1 وMALAT1 موارد جدیدی را نشان داده و سوالات جدیدی را ایجاد می کند. به عنوان مثال، نویسندگان جهش پرتکرار در یک منطقه غیر کد کننده ژن سرکوب گر اصلی تومور TP53 و جهش های کم تکرار را در مناطق غیر کد نویسی تلومراز ژن TERT مشاهده کرده اند؛ که منجر به بیان بیش از حد آنزیم تلومراز (که به سلول های توموری کمک می کند تا به طور غیرقابل کنترل تقسیم شوند) می شود.
بررسی سرطان تومورهای پیشرفته تر (متاستاتیک) اگرچه این مطالعه نتوانسته است وجود سایر مناطق غیر کدشده را رد کند، اما قاطعانه نشان می دهد که این نوع جهش شایع نیست.
۳و۴- الكساندر و همكاران۳ و لی و همكاران۴ روی ناهنجاری های ژنومی تمرکز کردند. فرآیندهای مختلف، مانند مکانیسم های ترمیم DNA یا قرار گیری در معرض جهش زاهای محیطی، این الگوهای مشخص شده، ناهنجاری ها را ایجاد می کنند. اگر بخواهیم اثرات شناخته شده جهش وموارد جدید را کشف کنیم، مجموعه های بزرگ ژنومی مهم هستند. الکساندروف و همکاران، لی و همکاران ۹۷ نشانه آن را شناسایی کردند.
قابل توجه است؛ لی و همکارانش از اولین کسانی هستند که اثرقابل بازآرایی مربوط به انواع ساختاری (Svs) را کشف کردند.( بازآرایی مجدد قسمت های زیادی از ژنوم) این فرآیند برای شناسایی محل جهش یافته به دلیل تنوع و پیچیدگی Sv بسیار مشکل می باشد.
مقاله مرتبط: تفاوت تومور با سرطان چیست و تومور خوش خیم و بدخیم چه فرقی دارند؟
Sv واثرات آن
محققان از طریق یک سری مراحل جهش زیر گروهی، اثر ۱۶Sv را شناسایی کردند؛ مثلاً بین دو Sv پیوند های مکانیکی، حذف ها و وارونگی های متقابل را نشان می دهد (آخرین مورد آن شامل معکوس جهت گیری است از یک بخش از Sv). همچنین آن ها از نقش ۱۶ اثر سرطان آگاهی یافتند . جهش در برخی از ژن های ترمیم کننده DNA با علائم سرطان مشخص شد. به عنوان مثال: این کنسرسیوم دریافت که جهش در ژن CDK12 در دو قطعه DNA حاصل از تکثیر، و نوع کوتاه آنزیم ترمیم کننده، DNA، همزمان با یک نشانه جهش متمایز شامل توالی DNA به نام سایت های CpG رخ می دهد. در مجموع این نشانه های جدید، پایه و اساس درک مکانیسم های رشد سرطان و نقش قرار گرفتن در معرض جهش زا در این فرایند را تشکیل می دهند.
پروسه ی تکامل سرطان
این ایده که سرطان از طریق یک فرایند تکاملی توسعه می یابد، برای اولین بار در سال ۱۹۷۶ ارائه شد. از آن زمان به بعد تکامل سرطان از نظر جهش های تصادفی و انتخاب طبیعی بررسی شده است. سلول سرطانی دارای جهشی است که موجب افزایش سریع تعداد سلول ها می شود، به عنوان برجسته ترین کلون سلولی در جمعیت تبدیل می شود. این پدیده را clonal sweep می نامند که طی چرخه های مکرر در طول رشد سرطان رخ می دهد. تکامل سرطان به طور مؤثر با توالی مناطق مختلف تومور با گذشت زمان مورد بررسی قرار گرفت و آن را از یک بیوپسی منفرد بازسازی کردند .
۵- رویکردی که در مقاله پنجم توسط Gerstung و همکاران صورت گرفت. نویسندگان مفهوم زمان مولکولی را برای طبقه بندی جهش های کلونال و زیر کلونال معرفی می کنند. آن ها استدلال كردند كه جهش هاي زيركلونال، فقط در زير مجموعه سلول هاي تومور وجود دارد؛ بايد ديرتر در تحولات سرطان پديد آيند. آن ها بسته به اینکه آیا جهش قبل یا بعد از اینکه کلون تعداد نسخه هایش را افزایش دهد (افزایش تعداد نسخه های یک ژن یا ناحیه کروموزومی) طبقه بندی می کنند. جهش های کلونال را که در همه سلول های تومور وجود دارد.
محققان داده های تکاملی را از تومورهای متعدد جمع کردند و به آن ها امکان می دهند مسیرهای جهشی مشترک مانند پیشرفت APC-KRAS-TP53 را شناسایی کنند، که توالی معمولی را توصیف می کند که در آن جهش ها در سرطان روده بزرگ بوجود می آید.
کدام جهش ها زودتر رخ می دهند؟
Gerstung و همکاران. دریافتند که جهش هایی که بیشتر در یک سرطان معین اتفاق می افتد نیز زودتر رخ می دهد. به طور مشابه، اگر دستاوردهای نسخه کپی در نوع خاصی از سرطان بسیار عود کنند، تمایل دارند زودهنگام رخ دهند. به عنوان مثال افزایش تعداد نسخه در بخشی از کروموزوم ۵ در سرطان کلیه شایع است و تمایل به بروز زودهنگام در تکامل بیماری بوجود می آید.
آیا جهش ها تغییرپذیر هستند؟
سرانجام محققان دریافتند که نشانه های جهش حداقل ۴۰٪ تومورها با گذشت زمان تغییر می کنند. این تغییرات نشان دهنده نقش كاهش در مواجهه با محیط زیست در پیشرفت بیماری و افزایش فراوانی و شدت نقایص ترمیم DNA است. به طور کلی یافته های این گروه نشان می دهد؛ که جهش می تواند سال ها قبل از تشخیص سرطان رخ دهد، که پیامدهای آن در تشخیص زود هنگام و توسعه نشانگرهای زیستی است.
۶-در مقاله آخر، PCAWG Transcriptome Core Group و همكارانشان ۱۸۸ نمونه PCAWG كه با داده هاي رونوشتي مطابقت داشتند، استفاده كردند تا بتوانند به طور عملكردي تغييرات DNA و RNA را ربط دهند. این گروه بین صدها جهش DNA تک نوکلئوتیدی و بیان ژن های مجاور ارتباط برقرار کردند. با این حال، تغییرات تعداد کپی بزرگتر عامل اصلی تغییرات ژن در سلول های سرطانی بودند. جهش همچنین با تغییر در ساختار منطقه، مانند تشکیل منطقه جدید کد کننده پروتئین (اگزون) در منطقه غیر کد نویسی (یک اینترون) همراه بود.
نويسندگان همچنين فراواني اتصالات پل را مشخص كردند – پديده اي كه در آن دو ژن به واسطه يك قطعه DNA مداخله شده توسط DNA، يكپارچه مي شوند. سرانجام ، اگرچه ۸۷ از ۱۸۸ نمونه مورد تجزیه و تحلیل هیچ تغییری در سطح DNA نداشت؛ اما این گروه نشان داد که هر یک از این موارد دارای تغییر در سطح RNA هستند. با همه ی این مشاهدات قدرت تجزیه و تحلیل توالی RNA- و توالی DNA برای مطالعات سرطان را نشان می دهد.
دسترسی به این مقالات
این شش مقاله، همراه با مقالات همراهی که در جای دیگری منتشر می شوند (به سایت go.nature.com/3boajsm مراجعه کنید)، نقطه عطفی در سرطان و ژنومی ابر است. PCAWG با تمرکز روی استنتاج ها، با موفقیت در یک دهه مطالعات توالی سرطان که عمدتاً در مشاهدات ریشه دارد گسترش می یابد. شایان ذکر است اگرچه تحلیل های استنباطی نسبت به مطالعات توصیفی، نگاه عمیق تری به سرطان نشان می دهد؛ اما نتایج آن ها هنوز قطعی نمی باشد.
در دسترس بودن و کیفیت گسترده مجموعه داده های PCAWG مطمئناً موجی از بینش های بیولوژیکی و پیشرفت های روش شناختی را ایجاد می کند. ادغام با سایر مجموعه های داده های ژنومی عملکردی، به عنوان مثال در مورد سازماندهی سه بعدی ژنوم، بدون شک درک دیگری از دلایل و پیامدهای ناهنجاری های ژنتیکی را نیز فراهم می کند.
چرا در مطالعات بالینی محدودیت هایی وجود دارد؟
بزرگترین محدودیت مطالعات فعلی عدم وجود اطلاعات بالینی در مورد نتایج و معالجه بیماران است. چنین داده هایی به محققان امکان می دهد تغییرات ژنتیکی را که می تواند نتایج بالینی را پیش بینی کند، شناسایی کنند. خوشبختانه پروژه ای به نام کنسرسیوم بین المللی – ژنوم سرطان در زمینه سرطان شناسی ژنومی (ICGC-ARGO) موسسه بین المللی انجام می شود که چنین منبعی را برای بیش از ۱۰۰۰۰۰ نفر مبتلا به سرطان ایجاد می کند.
سرانجام PCAWG هزاران دانشمند را گرد هم آورد، تا در رسیدن به اهداف خود با یکدیگر همکاری کنند. تأثیر طولانی مدت این تلاش ها فقط به یافته های منتشر شده امروز محدود نمی شود بلکه همکاری هایی که شکل گرفته اند و تبادل دانشی که بین اعضای این کنسرسیوم جهانی محققان صورت گرفته گسترش خواهد یافت.