مروری بر پاتولوژی کاردیومیوپاتی دیلاته ؛ کاردیومیوپاتی دیلاته بیماری پیشرونده و معمولاً برگشتناپذیری است که موجب اختلال عملکرد سیستولیک (انقباضی) سراسری به همراه نارسایی میشود. اغلب در این بیماری، آریتمیهای بطنی و فوق بطنی، اختلالات سیستم هدایتی و ترومبوآمبولیسم وجود دارد. مرگ ناگهانی ممکن است خصوصاٌ در مراحل آخر بیماری اتفاق بیفتد. اصطلاح کاردیومیوپاتی دیلاته از نقطه نظر پاتولوژیک، در حالت کلی به پروسهای ایدیوپاتیک در غیاب هایپرتنشن طولانی مدت، مواجهه با سموم یا الکلیسم مزمن (کاردیومیوپاتی دیلاته ثانویه) اطلاق میگردد. بیشتر بیماران در زمان شروع بیماری، میانسال یا مسنتر هستند. بیماران جوان مبتلا به کاردیومیوپاتی دیلاته اغلب سابقهی فامیلی و استعداد ژنتیکی برای این بیماری دارند (کاردیومیوپاتی دیلاته فامیلی). نارسایی احتقانی قلب با اختلال عملکرد سراسری بطن چپ نیز در بیماران مبتلا به ایسکمی کرونری و بیماری دریچهای ممکن است رخ دهد که از کاردیومیوپاتی دیلاته تقلید میکند.
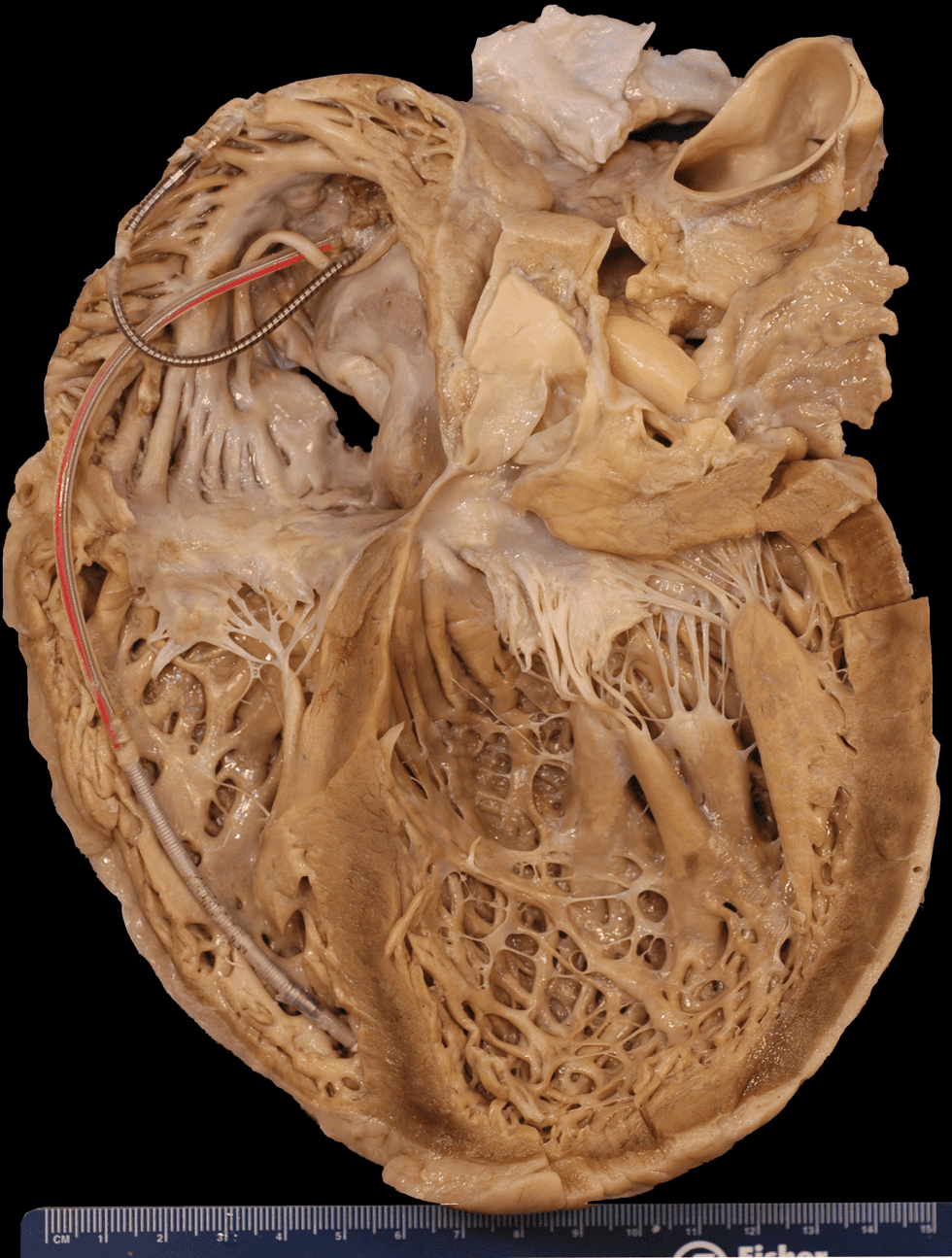
نمونهی قلب یک بیمار مبتلا به کاردیومیوپاتی دیلاته که در مراحل انتهایی نارسایی قلبی فوت کرده است. لیدهای دفیبریلاتور در قلب راست قرار دارند. بطنها در عین نرمال بودن ضخامت جدار بطن، دیلاته شدهاند؛ از این رو به نظر میرسد ضخامت جدار بطنها کمتر شده است. بطنها بیشتر از دهلیز دیلاته شدهاند.
کاردیومیوپاتی دیلاته با بزرگشدگی حفرهی بطن و اختلال عملکرد سیستولیک همراه با ضخامت نرمال بطن چپ (به شکل زیر رجوع کنید.) شناخته میشود. دیلاتاسیون و اختلال عملکرد بطن راست نیز ممکن است وجود داشته باشد؛ اما برای تشخیص ضروری نیست. کاردیومیوپاتی دیلاته پروسهای منتشر بوده و کاردیومیوسیتهای هر دو بطن درگیر میشوند. عملکرد دهلیز نیز کاهش مییابد. کتردیومیوپاتی دیلاته به عنوان یک بیماری اولیهی عضلهی قلب، علت مشخصی ندارد. درمان انتخابی این عارضه، پیوند قلب است.
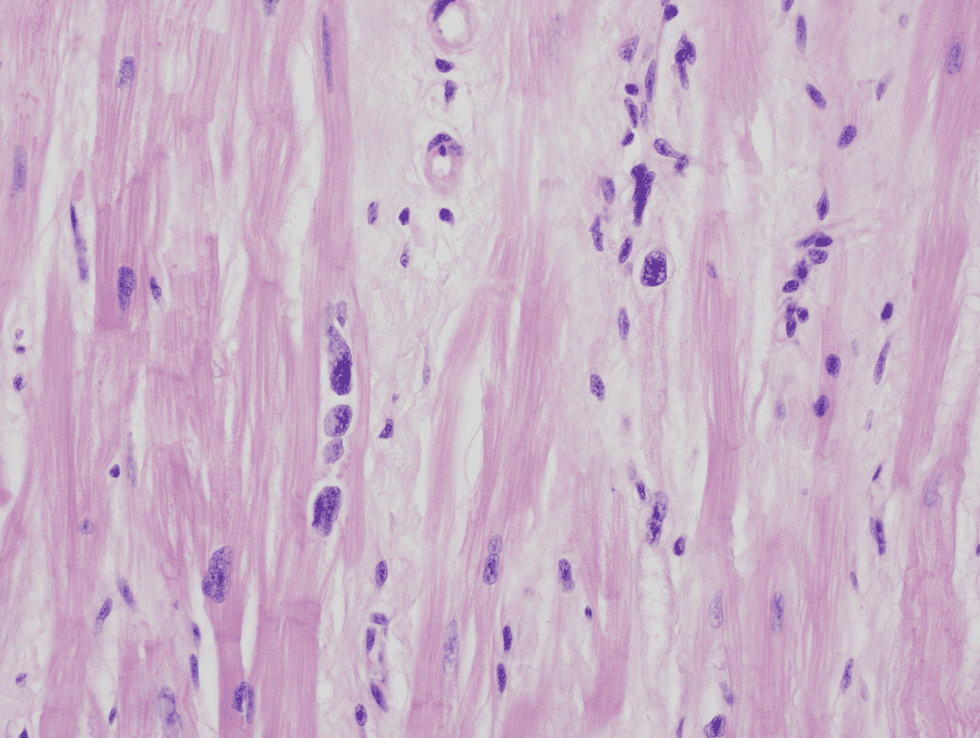
مقطع قلب به دست آمده از اکسپلنت قلبی در یک بیمار مبتلا به کاردیومیوپاتی که در مراحل انتهایی بیماری بوده است. به چندهستهای شدن کاردیومیوسیت توجه کنید. تغییرات اختصاصی نبوده و در نارسایی قلب ناشی از هر علتی میتوانند مشاهده شوند.
اتیولوژی
کاردیومیوپاتی دیلاته میتواند در انواع فامیلی، اولیه و بدون سابقهی فامیلی، یا ثانویه (مرتبط یا در اثر سایر بیماریها) طبقهبندی گردد. تقریباً ۲ نفر از هر سه بیمار، سابقهی فامیلی شناخته شده ندارند (کاردیومیوپاتی دیلاته اسپورادیک). قریب به ۱۵ درصد موارد اسپورادیک در زمینهی میوکاردیت مزمن بوده که در ادامه، موجب اسکار و نارسایی قلبی میشوند. ویروسهایی که باعث میوکاردیت میشوند، شامل کوکساکی ویروس، آدنوویروس، پاروویروس و ویروس نقص ایمنی انسان (HIV) هستند.
از جمله اتیولوژیها و موارد زمینهساز غیرالتهابی نیز الکلیسم، داروهای آنتراسیکلین، بلع فلزات، اختلالات سیستمیک و خودایمن و همچنین اختلالات میتوکندریایی را میتوان نام برد. گاهی اولیه یا ثانویه بودن کاردیومیوپاتی دیلاته چندان قابل تشخیص نیست؛ چنانچه تمییز میان ریسک فاکتور یا علت زمینهای بودن بسیاری از موارد مذکور نیز تاحدودی ناممکن است. کاردیومیوپاتی دیلاته فامیلی که حداقل عامل ۲۵% از موارد بیماری است، معمولاً اتوزومی غالب است؛ البته به دفعات کمتر به صورت وابسته به X مغلوب و همچنین با توراث میتوکندریایی نیز رخ میدهد.
اپیدمیولوژی
شیوع تخمینی کاردیومیوپاتی دیلاته ۱:۲۵۰۰ است. این بیماری از جمله شایعترین علل نارسایی قلبی میباشد. آمار تشخیص کاردیومیوپاتی دیلاته در اتوپسیها، هر ساله ۴.۵ مورد به ازای جمعیت ۱۰۰ هزار نفری است؛ حال آن که تشخیص بالینی بیماری، ۲.۴۵ مورد به ازای همین میزان از جمعیت میباشد.
کاردیومیوپاتی دیلاته به صورت بالینی ممکن است در طیف سنی گستردهای تظاهر یابد؛ اما این بیماری غالباً در دههی سوم یا چهارم زندگی بروز میکند.
پیشآگهی
نرخ بقای ۱۰ ساله در کاردیومیوپاتی کمتر از ۵۰ درصد است. اگرچه طبق گزارشهای موجود، نرخ بقای ۵ ساله و ۱۰ ساله با مراقبت حمایتی بهتر افزایش مییابد. کاردیومیوپاتی حوالی زایمان ممکن است در ۵۰ درصد بیماران برگشت داشته باشد؛ اما اغلب در حاملگی بعدی، مجدداً رخ میدهد.
در کاردیومیوپاتی دیلاته، میان بقا و تاکیآریتمی بطنی مکرر، ارتباط معکوس وجود دارد. این نوع تاکیآریتمی نیازمند درمان آنتیآریتمیک یا جاگذاری دفیبریلاتور کاردیوورتر قابل ایمپلنت و اتوماتیک (AICD) است.
ملاحظات تشخیصی
کاردیومیوپاتی دیلاته با افزایش اندازهی بطن چپ و کاهش کسر کوتاهشدگی، معمولاً به کمتر از ۲۵%، همراه است. باید خاطر نشان کرد اندازهگیری دقیق ابعاد پایان دیاستولی بطنی ناممکن بوده و اندازهگیریهای معاینات پس از مرگ، ممکن است ابعاد بطن را کمتر برآورد کنند.
کاردیومیوپاتی دیلاته معمولاً به هنگام شدید بودن علائم محدود کننده، تشخیص داده میشود؛ اما آریتمیها یا مرگ ناگهانی به ندرت به عنوان اولین تظاهر بالینی بروز مییابند. در مطالعات غربالگری فامیلی با اکوکاردیوگرافی، ممکن است بتوان بستگان بدون علامت یا با علائم خفیف بیمار را شناسایی کرد. آریتمیها در کاردیومیوپاتی دیلاته در مقایسه با کاردیومیوپاتی آریتموژنیک یا هیپرتروفیک، معمولاً تنها پس از شروع شاخص نارسایی قلبی واضح میشوند.
اصطلاح “کاردیومیوپاتی دیلاته خفیف”، بیماران با نارسایی قلبی پیشرفته بدون همودینامیک محدودکننده یا دیلاتاسیون قابل توجه بطن چپ را توصیف میکند. سابقهی فامیلی این حالت در بیش از۵۰ درصد از بیماران، مثبت است. تصویر بالینی و پیشآگهی کاردیومیوپاتی دیلاته خفیف، بسیار مشابه کاردیومیوپاتی دیلاته تیپیک است.
تشخیص افتراقی
تشخیص افتراقی بالینی و پاتولوژیک کاردیومیوپاتی دیلاته به معنای رد علل ثانویهی نارسایی قلبی است. از نظر پاتولوژیکی، یافتههای بافتشناسی غیر اختصاصی هستند (به بخش یافتههای میکروسکوپی رجوع کنید). از نظر ماکروسکوپی، قلب دیلاته و بزرگ شده در بیماری قلبی هایپرتنسیو طولانی مدت، بیماری دریچهای و بیماری کرونری شدید نیز مشاهده شده و این موارد باید رد شوند. در بیوپسی اندومیوکاردی نیز آملوئید، رسوب آهن و التهاب شاخص باید با رنگآمیزی معمول با رنگهای خاص رد گردند. بهعلاوه، قبل از تشخیص اختصاصی کاردیومیوپاتی دیلاته، شرح حال بالینی باید سایر علل نارسایی قلبی را رد کند.
کاردیومیوپاتی دیلاته در مقابل کاردیومیوپاتی ایسکمیک
تمییز کاردیومیوپاتی ناشی از بیماری شریان کرونری (کاردیومیوپاتی ایسکمیک) و کاردیومیوپاتی دیلاته اولیه از یکدیگر میتواند با مشکل مواجه شود. برخی بر این باورند که اگر بیماری شریان کرونری در همراهی با اختلال عملکرد سراسری بطن چپ، خارج از نسبت انسداد کرونری موجود باشد (به عنوان مثال، کسر تخلیهی کمتر از ۴۰% با تنگی ۵۰% یا کمتر در یک شریان پروگزیمال یا بیش از ۵۰% در یک شریان دیستال)، تشخیص کاردیومیوپاتی ایسکمیک داده میشود. با این حال، این تعریف کاردیومیوپاتی ایسکمیک، ناکارآمد بوده و در حال حاضر اصطلاح کاردیومیوپاتی ایسکمیک به بیماران مبتلا به بیماری کرونری شدید و اختلال عملکرد سراسری بطن چپ، معمولاً همراه با انفارکت ترانسمورال بهبود یافته، اطلاق میشود.
یافتههای ماکروسکوپی
اصلیترین علامت در یافتههای ماکروسکوپی اتوپسی، دیلاتاسیون بطن چپ معمولاً به اندازهی بیش از ۴ سانتیمتر است. کاردیومگالی معمولاً لازمهی تشخیص کاردیومیوپاتی دیلاته در نظر گرفته میشود. میانگین وزن قلب تقریباً ۶۰۰ گرم است. بزرگشدگی قلب در برخی بیماران مبتلا به کاردیومیوپاتی دیلاته بسیار کم بوده و تشخیص باید مبتنی بر زمینههای بالینی صورت گیرد. در شرایط معمول، دیلاتاسیون ۴ حفرهای دیده میشود که در بطن بیشتر از دهلیزهاست. در بیماران با سابقهی فیبریلاسیون دهلیزی نیز ممکن است دیلاتاسیون دهلیزی وجود داشته باشد.
بهترین روش برای ارزیابی دیلاتاسیون بطن چپ در اتوپسی، اندازهگیری حجم حفره در سطح عضلات پاپیلری در برش عرضی است. دیلاتاسیون همزمانِ بطن راست، به نمای کروی تیپیک قلب منتج میشود.
در مطالعهی صورت گرفته بر ۶۴ اکسپلنت قلبی با تشخیص بالینی کاردیومیوپاتی دیلاته، ۹ قلب نشان از سایر بیماریها داشتند؛ به این معنا که احتمالاً تشخیص بالینی نادرست بوده است. در این مطالعه، ۵۵ بیماری که با استناد به یافتههای پاتولوژیک مبتلا به کاردیومیوپاتی دیلاته بودند، اکثراً مرد بوده (۶۵%) و میانگین سنشان در زمان پیوند ۴۸ سال بود. ۱۶% این افراد سابقهی فامیلی کاردیومیوپاتی داشتند. یافتههای پاتولوژیک در این ۵۵ قلب شامل زیرمجموعههای متعدد بود: ۳۸ مورد دیلاتاسیون تیپیک ۴ حفرهای، ۵ مورد اختلال بطن چپ، ۴ مورد تغییرات بافتی یا ماکروسکوپی حداقل (کاردیومیوپاتی دیلاته حداقل) و ۳ مورد الگوی میوکاردیت بهبودیافته داشتند.
ضخامت جدار بطن چپ در مقایسه با کاردیومیوپاتی هایپرتنسیو توأم با نارسایی، اغلب نرمال است. همچنین ممکن است در اثر اختلال عملکرد عضلهی پاپیلری که به طور ثانویه به دیلاتاسیون بطنی و تغییرات در شکل جدار بطن رخ میدهد، نارسایی میترال بروز یابد. در مقایسه با این حالت، رگورژیتاسیون تریکوسپید در اثر دیلاتاسیون حلقوی شکل ایجاد میشود.
ترومبوز جداری در بیمارانی که داروی ضدانعقاد دریافت نمیکنند، شایع است. در حدود ۱۰ درصد موارد، پلاکهای فیبروزی اندوکاردیال بطنی، احتمالاً به طور ثانویه و در اثر ترومبوز، دیده میشود. فیبروز اندوکاردیال تکه تکه یا منتشر خفیف نیز خصوصاً در نزدیکی راه خروجی بطن، به طور مکرر دیده شده و به نظر میرسد ناشی از دیلاتاسیون قلبی است.
یافتههای میکروسکوپی
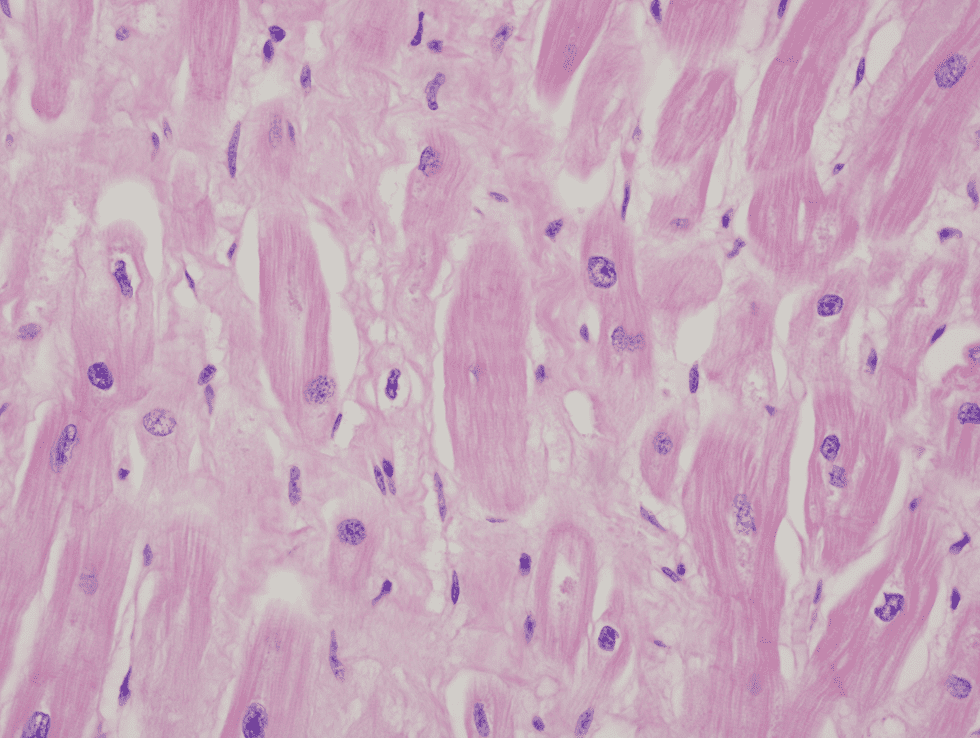
مقطع قلب به دست آمده از اکسپلنت قلبی در یک بیمار مبتلا به کاردیومیوپاتی که در مراحل انتهایی بیماری بوده است. فیبروز بینابینی کانونی دیده میشود. تغییرات غیراختصاصی بوده و در نارسایی قلبی ناشی از هر علتی دیده میشوند.
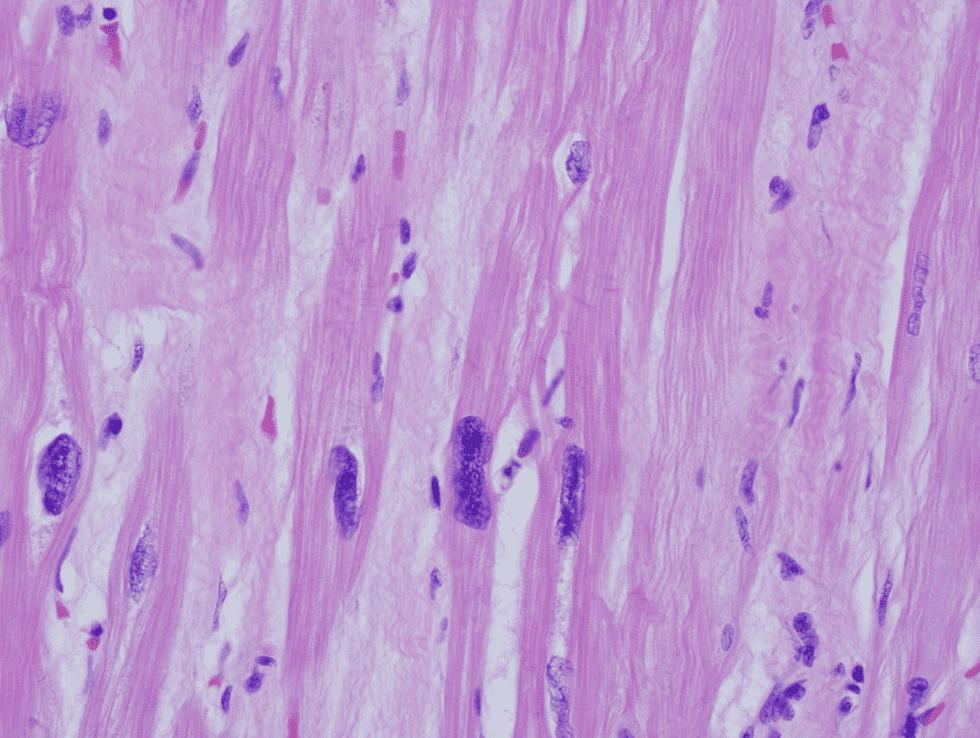
مقطع قلب به دست آمده از اکسپلنت قلبی در یک بیمار مبتلا به کاردیومیوپاتی که در مراحل انتهایی بیماری بوده است. به واریاسیون در سایز هستهها توجه کنید. تغییرات غیراختصاصی بوده و در نارسایی قلبی ناشی از هر علتی دیده میشوند.
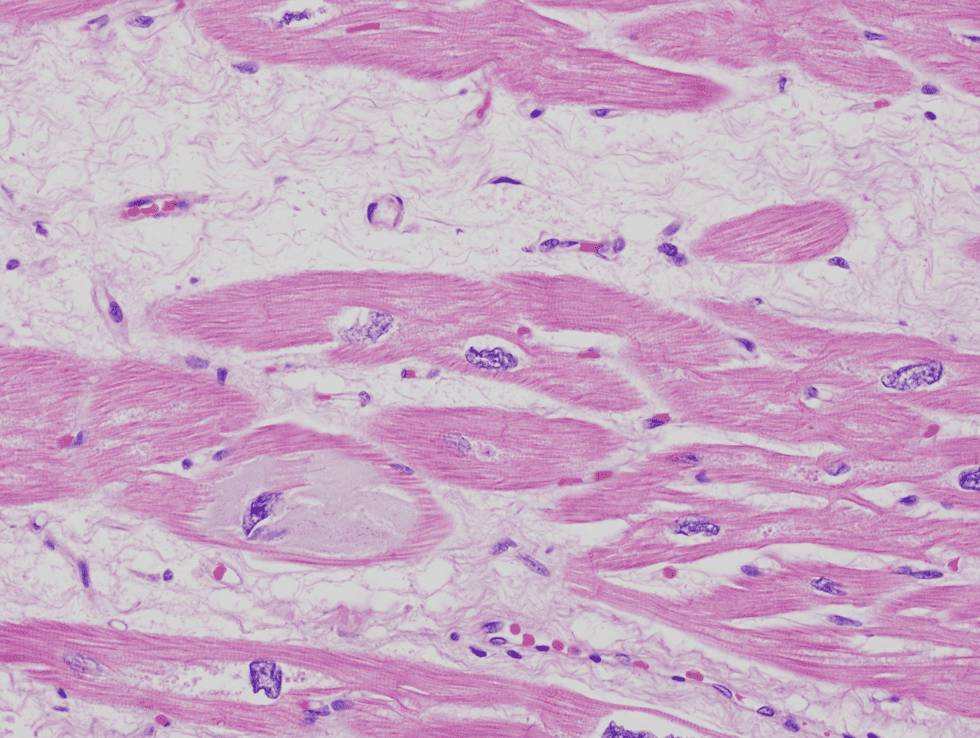
مقطع قلب به دست آمده از اکسپلنت قلبی در یک بیمار مبتلا به کاردیومیوپاتی که در مراحل انتهایی بیماری بوده است. به تجمع داخل سلولی مادهی با ساختمان غیرمشخص توجه کنید (دژنراسیون بازوفیلیک). تغییرات غیراختصاصی بوده و در نارسایی قلبی ناشی از هر علتی دیده میشوند.
ایمونوهیستوشیمی
تکنیکهای ایمونوهیستوشیمیایی (IHC) برای تشخیص کاردیومیوپاتی دیلاته کاربردی نیستند. ایمونولوکالیزاسیون پروتئینهای اسکلت سلولی و پروتئینهای سارکومریک در قلب اکسپلنت شدهی بیماران مبتلا به کاردیومیوپاتی دیلاته، نشان دهندهی پراکندگی غیرطبیعی آنهاست. مقدار توبولین و دسمین نیز به صورت افزایش یافته و پراکندگی آنها به طور نامنظم گزارش شده است. تیتن، یکی از اعضای خانوادهی اسکلت سارکومری، کاهش داشته که این کاهش خصوصاٌ در نواحی دچار کمبود مادهی انقباضی بیشتر میباشد. کانکسین-۴۳ نیز کاهش داشته است. افزایش مرگ سلولی و آپوپتوز میوسیتها به همراه رژنراسیون میوسیتها با بیان آنتیژن هستهای تکثیر سلولی (PCNA) و Ki-67 هم مشاهده گردید.
ایمونولیبلینگ فوق ساختاری در بیماران حامل جهشهای لامین-A، حاکی از عدم وجود پوشش هستهای در بیماران حامل جهشهای ژن لامین A/C تحت عنوان LMNA است. در موارد مرتبط با دیستروفینِ کاردیومیوپاتی دیلاته (تا حدود ۶% مردان مبتلا به بیماری)، مطالعات ایمونوهیستوشیمیایی و مولکولی برای شناسایی نقایص پروتئینی و ژنی الزامی است.
ژنتیک
فرم اتوزومی غالب کاردیومیوپاتی دیلاته به طور اولیه به دنبال جهشهایی در ژنهای کدکنندهی پروتئینهای اسکلت سلولی رخ میدهد. به میزان کمتر نیز این بیماری در اثر جهشهای درگیرکنندهی پروتئینهای سارکومر، غشای هستهای (شامل پروتئینهای فعالکنندهی رونویسی) و دیسک اینترکاله بروز مییابد. شایعترین جهشهای شناسایی شده، جهشهای ژن LMNA است. این ژن، پروتئینهای پوشش هستهای تحت عنوان لامین A و لامین C را کد میکند. بیماران حامل این جهش اغلب بلوک دهلیزی بطنی دارند.
همچنین ژن وابسته به X مسبب دیستروفی عضلانی امری دریفوس، اِمِرین (یکی دیگر از پروتئینهای لامین هستهای)، ممکن است موجب کاردیومیوپاتی دیلاته گردد. سایر بیماریهای وابسته به X مرتبط با کاردیومیوپاتی دیلاته شامل دیستروفیهای عضلانی (همچون بکر و دوشن) است. سطح کراتین کیناز سرم این بیماران اغلب بالا است. کاردیومیوپاتی دیلاته در بیماران مبتلا به میوپاتیهای میتوکندریایی و اختلالات متابولیک ارثی (مانند هموکروماتوز) نیز ممکن است رخ دهد.
شیوع شناسایی پروتئینهای سارکومریک در کاردیومیوپاتی دیلاته، رو به افزایش است. این پروتئینها شامل اکتین آلفای قلبی؛ آلفا تروپومیوزین؛ تروپونین T، I و C قلبی؛ زنجیزهی سنگین میوزین بتا و آلفا، میوزین متصل شونده به پروتئین C، پروتئین LIM عضله؛ آلفا-اکتینین ۲؛ ZASP و تیتین است. در برخی خانوادههای حامل جهشهای سارکومریک، فنوتیپهای دیلاته و هیپرتروفیک همپوشانی داشته که نشان دهندهی انعطافپذیری این ژنها و همچنین اختلاف گهگاه ژنوتیپ با فنوتیپ است. این یافتهها تأکید بر اهمیت تعریف کاردیومیوپاتی با مورفولوژی و نه ویژگیهای ژنتیکی دارند.