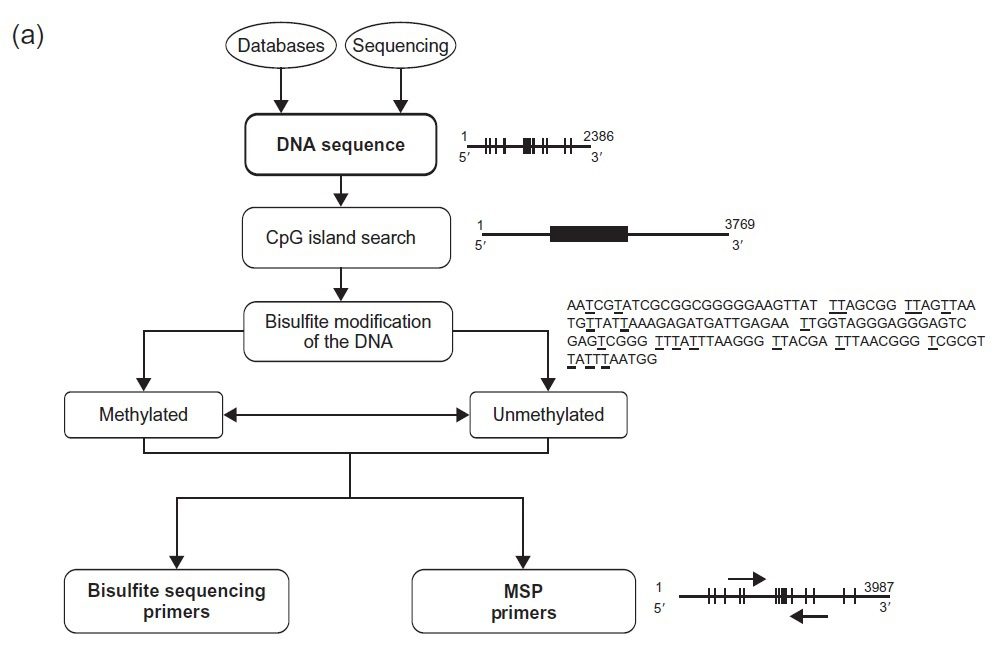
تکنیک PCR اختصاصی متیلاسیون (Methylation specific PCR= MSP)، متدی ساده، سریع و مقرون به صرفه به منظور آنالیز وضعیت متیلاسیون DNA است که تقریبا میتواند روی هر گروه از CpGهای داخل یک جزیره CpG که در پروموتر ژنها واقع است، انجام بگیرد. این تکنیک در سال ۱۹۹۶ شناسایی شده و یکی از کاربردهای توالییابی بی سولفیت میباشد.
مقالات مرتبط:
متیلاسیون DNA
متیلاسیون DNA، از مهمترین تغییرات اپیژنتیکی است که در سلولهای یوکاریوتی رخ میدهد و تقریبا یک درصد از بازهای DNA انسان تحت تاثیر آن قرار میگیرند. این پدیده نقش مهمی در تنظیم بیان ژن، حفظ یکپارچگی ساختار ژنومی و حکگذاری ژنی دارد. در طی متیلاسیون، افزودن گروه متیل پس از همانندسازی و غالبا به کربن شماره ۵ حلقه پریمیدین سیتوزین انجام میگیرد، که منجر به تولید ۵-متیل سیتوزین میگردد. این فرایند تنها در بازهای سیتوزینی که در سمت ۵’ گوانوزین قرار داشته باشند و به نام دینوکلئوتیدهای CpG خوانده میشوند، رخ میدهد. گروه متیل در خارج از مارپیچ دورشتهای DNA قرار دارد. از این رو، متیلاسیون سیتوزینها مانع از جفت شدن بازها نمیگردد و ۵-متیل سیتوزین همانند سیتوزین عادی میتواند با گوانین جفت شود.
متیلاسیون سیتوزین توسط آنزیم DNA متیل ترنسفراز (DNMT) صورت میگیرد و طی آن انتقال گروه متیل از S-adenosylmethionine (دهنده گروه متیل) به سیتوزین (گیرنده متیل) انجام میشود. انسانها، ۳ نوع DNMT عملکردی دارند. پروتئین چهارم که DNMT3L نام دارد، به هدفگیری توالی مناسب توسط متیلازها کمک میکند و پروتئین پنجم، DNMT2، علیرغم ساختار مشابهش با DNA متیل ترانسفرازها، یک آنزیم متیلهکننده RNA است.
حضور متیل سیتوزین در پروموتر، موجب تغییر در اتصال فاکتورهای رونویسی و سایر پروتئینها به مولکول DNA شده و درنتیجه پروتئینهای methyl CpG-binding (MeCpG) و هیستون داستیلازها به محل فراخوانده میشوند. این پروتئینها در تنظیم بیان ژن و ساختار کروماتین نقش دارند و باعث فشردگی کروماتین در اطراف محل شروع رونویسی ژن میشوند. MeCpG ها همچنین دارای نقشی مهم در حافظه اپیژنتیکی هستند. سیتوزینهای متیلهشده در ارتباط با DNMTها، پروتئینهای متصل شونده به متیل، هیستون دِاستیلازها و پروتئینهای سرکوبکننده رونویسی هستند که مجموعا به همراه یکدیگر ساختار کروماتینی مهارکننده رونویسی را تشکیل میدهند. بررسی وضعیت متیلاسیون سیتوزینها، با قرار دادن DNA در معرض سدیم بیسولفیت و آنالیز نتایج انجام میشود و در ادامه به آن خواهیم پرداخت.
دینوکلئوتیدهای CpG در ژنوم، کمترین فراوانی را نسبت به سایر دینوکلئوتیدها در ژنوم انسان دارند. علت این موضوع از دست رفتنشان در طی تکامل است که به تمایل متیل سیتوزینها به دآمیناسیون خودبهخودی به تیمینها مرتبط میباشد. تنها نواحی کوچکی از DNA به نام جزایر CpG حاوی دینوکلئوتیدهای CpG است که استثنا محسوب میشود. این جزایر در نواحی پروموتر بسیاری از ژنها یافت میشود و فراوانی آنها نیز از طریق محاسبات ریاضی قابل پیشبینی است. هر جزیره CpG، کشیدگی ۰.۵ تا ۴ کیلوبازی DNA با محتوای G:C بزرگتر از ۵۵ درصد است. این جزایر، حاوی حدودا ۲۰ درصد کل دینوکلئوتیدهای CpG هستند و با نواحی پروموتر تقریبا نیمی از ژنها مرتبط میباشند.
پروموترهای حاوی جزایر CpG، هم در شرایط فعال بودن ژن و هم در حالت غیرفعال بودن آن، شدیدا از متیلاسیون محافظت میشوند. این در حالی است که دی نوکلئوتیدهای CpG غیرمرتبط با جزایر CpG شدیدا متیله میگردند. علت این موضوع، احتمالا فشار انتخابی وارد آمده بر جزایر واقع در پروموترها، در جهت حفظ فعالیت پروموتر بوده است. گاها جزایر CpG در ژنوم دچار متیلاسیون DNA میشوند و این موضوع، به طرز غیرقابل اجتنابی، باعث خاموش شدن رونویسی ژن میگردد؛ مثلا در کروموزوم X غیرفعال یا ژنهای حکگذاریشده.
متیلاسیون DNA در رشد طبیعی پستانداران حیاتی بوده و با نقشپذیری ژنی، غیرفعالسازی رونویسی کروموزوم X و پیری در ارتباط میباشد. اکثریت ژنهای پستانداران (در حدود ۷۰٪ آنها) متیله شده اند. متیلاسیون سیتوزینها در DNA ژنومی، در تنظیم بیان ژن نقش اساسی دارد. سه مکانیسم اصلی مرتبطکننده متیلاسیون به سرطان عبارتند از: خاموش کردن رونویسی ژنهای سرکوبکننده تومور در هایپرمتیلاسیون نواحی پروموتر، هایپومتیلاسیون پروتوانکوژنها که منجر به عدم توانایی مهار آنها میگردد؛ و هایپومتیلاسیون کل ژنوم (نئوپلازی) که منجر به افزایش میزان جهشها و ناپایداری کروموزوم میشود. آنالیز هایپرمتیلاسیون پروموتر و هایپومتیلاسیون کل ژنوم، میتواند به درک ما از سرطان کمک کرده و به عنوان مارکر مولکولی در زمینههای تشخیصی به کار رود.
تشخیص متیلاسیون DNA
متیلاسیون DNA ژنومی، از آنجایی که مکانیسمی به منظور کنترل بیان ژن است، امروزه به میزان زیادی مورد توجه دانشمندان میباشد. متدهای فراوانی به منظور آنالیز متیلاسیون DNA توسعه یافته اند. تعیین سطح متیل سیتوزین موجود در کل ژنوم میتواند از طریق تکنیکهای جداسازی با کارایی بالا و یا روشهای آنزیمی و مکانیکی انجام بگیرد. روشهای دسته دوم حساسیت کمتری نسبت به دسته اول دارند و غالبا رزلوشن آنها وابسته به محلهای برش اندونوکلئاز است. علیرغم این نقایص، روشهای آنزیمی و شیمیایی به علت عدم نیاز به تجهیزات پیچیده و گرانقیمت، امروزه نیز پرکاربرد هستند.
بررسی الگوهای متیلاسیون اختصاصی بافت می تواند از طریق متدهای وابسته به هیبریداسیون in situ و استفاده از آنتیبادیهای آنتی-متیل سیتوزین لیبلشده، انجام شود. متیلاسیون DNA میتواند در کروموزومهای متافازی، هتروکروماتینها و یوکروماتینهای موجود در داخل سلول نیز آنالیز شود که در این صورت، میتواند امکان تشخیص تفاوتهای میان سلولهای نرمال و سرطانی موجود در نمونههای یکسان را فراهم آورد. استفاده از microarray های اختصاصی متیلاسیون نیز از تکنیکهای جدید است که امکان بررسی وضعیت متیلاسیون چندین ژن مختلف را در تنها طی یک آزمایش پدید میآورد.
امروزه بسیاری از متدهای موجود برای مطالعه تغییرات وضعیت متیلاسیون پروموترهای حاوی جزیره CpG وابسته به ایجاد تغییر در DNA تحت تاثیر بیسولفیت و در نتیجه فیکس شدن الگوی متیلاسیون، می باشند. سپس تکثیر توسط PCR انجام میگیرد و امکان تمایز اللهای متیله شده و متیله نشده از یکدیگر فراهم میگردد. بدون اعمال بی سولفیت، الگوی متیلاسیون در طی واکنش تکثیر PCR حفظ نخواهد شد. البته استفاده از بی سولفیت، مضراتی نیز دارد و آن تجزیه DNA و کاهش محصول است.
بررسی متیلاسیون تا مدتها توسط هیبریداسیون ساترن و اندونوکلئازهای اختصاصی متیلاسیون انجام میشد. این متدها، بر پایه ناتوانی آنزیمهای محدودکننده حساس به متیلاسیون در برش سیتوزینهای متیله شده موجود در جایگاه تشخیص میباشند. برخی از اندونوکلئازها، برش DNA دورشتهای در محل جایگاه تشخیص خود را زمانی انجام میدهند که سیتوزینها فاقد متیلاسیون باشند؛ در حالیکه برخی دیگر، این کار را بدون در نظر گرفتن حضور یا نبود ۵-متیل سیتوزین انجام میدهند.
در این تکنیکها، DNA ژنومی توسط اندونوکلئازهای حساس به متیلاسیون برش داده شده، در ژل آگارز الکتروفورز و به پروبهای اختصاصی DNA متصل میگردد. ناتوانی آنزیم در برش توالیهای متیلهشده، منجر به تولید قطعاتی با طول بلندتر خواهد شد که نشانگر دینوکلئوتید CpG متیلهشده میباشد. در نتیجه در الکتروفورز، نوارهایی با طولی بزرگتر از حد موردانتظار ظاهر میشود.
مشکل اساسی که در رابطه با اندونوکلئازهای اختصاصی متیلاسیون و ساترن بلاتینگ وجود دارد این است که تنها سیتوزینهایی میتوانند مورد بررسی قرار بگیرند که بتوانند توسط اندونوکلئازهای اختصاصی متیلاسیون موجود، تشخیص داده شوند (به عنوان مثال، HpaII (CCGG) و HhaI (GCGC)). به علاوه، استفاده از این متدها مناسب آنالیزهای متیلاسیونی که روی کل ژنوم انجام میگیرد و نیز تکنیکهای marker discovery است، اما در بررسی وضعیت متیلاسیون جایگاههای CpG اختصاصی چندان مفید عمل نمیکند. این تکنیکها، علیرغم سادگی، سرعت و حساسیت بالا، نیازمند مقادیر بالایی از DNA با کیفیت هستند. میتوان با استفاده از PCR، نقص گفته شده را برطرف نمود.
در صورت استفاده از PCR، تجزیه مولکولهای DNA با اندونوکلئازهای حساس به متیلاسیون باید پیش از تکثیر توسط PCR انجام بگیرد. زیرا متیلاسیون سیتوزین نباید پس از PCR حفظ شود و ۵- متیل سیتوزین باید به سیتوزین تبدیل گردد. پرایمرها نیز باید اختصاصی نواحی بالادست و پایین دست جایگاه تشخیص اندونوکلئاز حساس به متیلاسیون باشند. سپس DNA شکسته شده تحت تاثیر آنزیم، توسط پرایمرها تکثیر میشود. در صورت متیله بودن جایگاه تشخیص، DNA الگو برش داده نمیشود و شاهد تولید محصول PCR خواهیم بود. در صورتی که جایگاه تشخیص غیرمتیله باشد، از آنجایی که توالی الگو به قطعاتی تقسیم میشود، محصولی ایجاد نخواهد شد. حتی اگر DNA الگو در جایگاه تشخیص خود کاملا غیرمتیله باشد، به علت حساسیت بالای PCR، وجود نقص در برش در مقادیر اندک نیز میتواند به نتایج مثبت کاذب منجر شود. پس تفسیر نتایج این تکنیک باید با احتیاط صورت گیرد.
متدهای آنالیز متیلاسیون DNA با استفاده از PCR
تکنیکهای وابسته به PCR، به منظور مطالعه وضعیت متیلاسیون نواحی خاصی از DNA، مانند جزایر CpG موجود در پروموترها مورداستفاده قرار میگیرند. دو روش اصلی به این منظور وجود دارد:
- عدم اعمال بی سولفیت: در این روش، DNA ژنومی به صورت مستقیم و بدون هیچ گونه تغییری مورداستفاده قرار میگیرد و طی آن از اندونوکئازهای محدودکننده حساس به متیلاسیون در ترکیب با تکنیکهای تشخیصی مانند ساترن بلاتینگ و PCR استفاده میشود و پیشتر به آن پرداختیم.
- اعمال بی سولفیت: تمام متدهای مرتبط با بی سولفیت نیازمند تکثیر DNA تغییر یافته تحت تاثیر این ماده توسط تکنیک PCR میباشند. از جمله روشهای این نوع از آنالیز، methylation-specific PCR (MSP) است.
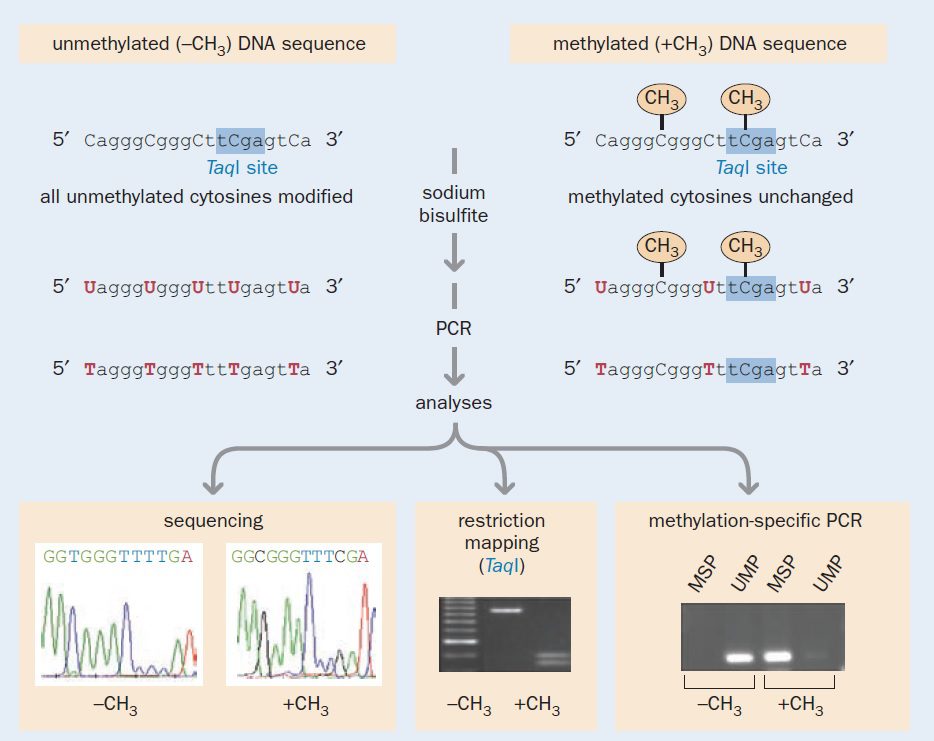
معرفی توالی یابی سدیم بی سولفیت توسط Frommer و همکارانش در سال ۱۹۹۲، مطالعه وضعیت متیلاسیون سیتوزینها را در بسیاری از آزمایشگاهها ممکن ساخت و زمینهساز پیشرفتهای فراوانی شد که در زمینه تحقیقات متیلاسیون DNA انجام گرفته است. استفاده از سدیم بی سولفیت امکان تغییر DNA ژنومی و فیکس کردن الگوی متیلاسیون را فراهم کرده است. بررسی متیلاسیون DNA به این طریق میتواند موانع موجود در آنالیز آنزیم محدودکننده را دور بزند و امروزه به طور گستردهای مورد استفاده است.
در شرایط کنترل شده، منجر به دآمیناسیون سیتوزینهای متیله نشده در موقعیت ۴ آنها و تبدیل آنها به یوراسیل و در نتیجه ایجاد تفاوت در توالی میگردد. در حالیکه، سیتوزینهای متیلهشده (۵-متیل سیتوزین) به دآمیناسیون در اثر سدیم بی سولفیت مقاوم بوده و بدون تغییر باقی میمانند. در صورتی که DNA تغییریافته، توالییابی شده و یا توسط واکنش PCR تکثیر شود، یوراسیلها، به تیمین تبدیل خواهند شد و سیتوزینهای متیلهشده به صورت سیتوزین باقی خواهند ماند. به این ترتیب، متیلاسیون سیتوزین که تغییری اپیژنتیکی است، به صورت تفاوت در ساختار اولیه DNA (ترکیب بازها) نمایان میشود.
در توالییابی بیسولفیت، محصولات تکثیرشده کلون و توالییابی می شوند. با مقایسه توالی DNA پیش و پس از تیمار با بی سولفیت، تشخیص سیتوزینهای متیله ممکن میگردد؛ به این نحو که تمام سیتوزینهای non-CpG موجود غیرمتیله بوده و به تیمین تبدیل میشوند و تنها ۵-متیل سیتوزینهای موجود در CpGها هستند که در توالی یابی به صورت سیتوزین باقی میمانند. این گونه است که مولکولهای DNA متیله شده و متیله نشده از یکدیگر تمایز پیدا میکنند.
علاوه بر توالی یابی، متدهای بسیاری به منظور تشخیص تغییرات ایجاد شده در نتیجه تیمار DNA با سدیم بی سولفیت موجود است. وضعیت توالیهای مشخص CpG در ژنوم میتواند توسط تجزیه با آنزیم محدودکننده و یا PCR (تکنیک MSP) بررسی شود. تغییر باز A به باز T میتواند منجر به ایجاد جایگاه تشخیصی خاص و یا از بین رفتن آن شود. به عنوان مثال، در صورتی که باز C غیرمتیله باشد، جایگاه تشخیص TCGA که متعلق به TaqI است، تخریب میشود.

Methylation-specific PCR (تکنیک PCR اختصاصی متیلاسیون)
methylation specific PCR) MSP) در سال ۱۹۹۶ توسط Herman و همکارانش شناسایی شد و به طور گستردهای در مطالعه جزایر CpG پروموترها مورداستفاده قرار میگیرد. این تکنیک از دو بخش تشکیل شده است: ۱) تبدیل سیتوزینهای متیلهنشده به یوراسیل تحت تاثیر سدیم بیسولفیت، در شرایطی که سیتوزینهای متیلهشده بدون تغییر باقی میمانند؛ و ۲) شناسایی تفاوتهای توالی القاشده در اثر بیسولفیت تحت تاثیر PCR، با استفاده از پرایمرهای اختصاصی هر دو نوع DNA متیله و غیرمتیله.
تفاوت در وضعیت متیلاسیون که با واکنش بی سولفیت نشانهگذاری میشود، میتواند با دقت بالایی توسط تکنیک MSP نیز مورد سنجش کمی قرار بگیرد. اساس این موضوع آن است که پس از قرار دادن توالیهای حاوی CpG تحت تاثیر سدیم بی سولفیت، قطعا اللهایی که CpGهایشان متیله هستند نسبت به آنهایی که غیرمتیله هستند، توالی متفاوتی را حاصل خواهد کرد. در این حالت، از دو مجموعه پرایمر متمایز برای توالی موردنظر استفاده میشود، به نحوی که سیتوزینهای متیله و غیرمتیله در DNA تغییر یافته در اثر بی سولفیت از یکدیگر تشخیص داده شوند. پرایمر متیلهنشده (UMP)، تنها مولکول DNA ای را که تحت تاثیر سدیم بی سولفیت تغییر یافته است را تکثیر میکند. این در حالی است که پرایمر متیلهشده (MSP)، اختصاصی DNA متیله شده تغییر یافته تحت تاثیر سدیم بی سولفیت میباشد.
هنگامی که مجموعه پرایمری مورد استفاده قرار گیرد که مکمل CpG متیله شده بوده و غیرمکمل با CpG متیله نشده همان توالی باشد، تنها الل حاوی CpG متیله شده باید تکثیر شود، و برعکس. تفسیر نتایج نیز آسان است: در صورتیکه اندازه محصولات PCR که روی ژل آگارز دیده میشود، طبق انتظار باشد، بسته به نوع پرایمر مورد استفاده، متوجه میشویم که نمونه حاوی الل متیله شده و یا غیرمتیله ژن است. کاری که معمولا انجام میشود، استفاده از دو جفت پرایمر اختصاصی توالیهای متیله شده و متیله نشده برای همان ژن و الکتروفورز هر دو به صورت ساید بای ساید به منظور مقایسه است.
پروفایل متیلاسیون کل ژنوم میتواند با آنالیز DNA تیمارشده با بیسولفیت که روی microarray های الیگونوکلئوتیدی مخصوص قرار دارد و حاوی پروبهایی اختصاصی توالی نرمال یا تغییریافته است نیز، تهیه شود. با تکنیکهای توالی یابی بی سولفیت و pyrosequencing هم میتوان متیلاسیون را آنالیز کرد. تمامی این تکنیک ها مبتنی بر PCR هستند.
مهمترین مزیت MSP نسبت به سایر متدهای آنالیز متیلاسیون DNA، سادگی آن است. در مقایسه با ساترن بلاتینگ با استفاده از آنزیمهای محدودکننده حساس به متیلاسیون، MSP نیازمند مقادیر کمتری از DNA است، معمولا نیاز به استفاده از ایزوتوپها نیست و تمام CpGها، صرف نظر از توالیهای اطرافشان میتوانند به این نحو مورد بررسی قرار بگیرند. همچنین تفسیر نتایج بسیار آسانتر است. مزیت MSP نسبت به توالی یابی بی سولفیت آن است که نیاز به کلونینگ و توالی یابی ندارد و درنتیجه به جای اینکه چندین روز زمان بگیرد، در عرض یک یا دو روز به انجام میرسد.
تطبیقپذیری MSP با انواع کاربردها، حساسیت بالا(قدرت تشخیص یک الل متیلهشده در ازای ۱۰۰۰ الل متیلهنشده)، سرعت و مقرون به صرفه بودن آن، این تکنیک را به روشی مناسب در آنالیز نمونههای بالینی با مقادیر ورودی بالا تبدیل کرده و استفاده از آن را برای محققانی که تعداد زیادی از نمونههای بالینی را به صورت همزمان بررسی میکنند، مناسب نموده است. از جمله کاربردهای بالینی MSP، آنالیز بیماران دارای نقص در متیلاسیون DNA، بررسی وضعیت هر گونه تولی DNA مانند ژنهای ویروسی و ژنهای حکگذاری شده وابسته به X و یا اتوزومی و نیز بررسی میزان پاسخ بیماران سرطانی به داروهای آلکیلهکننده میباشد. با استفاده از MSP محققان توانستهاند هایپرمتیلاسیون آنزیم MGMT را که در ترمیم DNA نقش دارد و هایپرمتیلاسیون و غیرفعال شدن ژنهای سرکوب کننده تومور را تعیین کنند.
البته MSP محدودیتهایی نیز دارد که حتما باید مدنظر قرار گیرند. MSP تکنیکی کمی نیست و تنها حضور یا عدم حضور متیلاسیون را در ناحیه موردنظر تعیین میکند. علت این موضوع آن است که تکنیکهای توالی یابی بی سولفیت، امکان بررسی کیفی وضعیت متیلاسیون ۵-متیل سیتوزین موجود در امپلیکون را میان توالیهایی که پرایمر به آنها متصل میشود، فراهم میآورند. در نتیجه این تکنیکها نیازمند پرایمرهای اختصاصی توالیهای تغییریافته تحت تاثیر بیسولفیت هستند، نه آنهایی که لزوما اختصاصی DNA متیله شده و یا متیلهنشده میباشند.
همچنین این امکان وجود دارد که مرحله تکثیر PCR که به دنبال اعمال بی سولفیت رخ میدهد، علاوه بر نتایج حقیقی، نتایج کاذب را نیز به صورت نمایی تکثیر کند. از این رو، میزان موفقیت تاثیر بی سولفیت باید با استفاده از کنترلها، در تمام آزمایشات مانیتور شود تا از درست بودن نتایج اطمینان حاصل کنیم. از جمله حالات ممکن شامل دناتوراسیون ناقص مولکول DNA و تغییر یافتن متیلسیتوزینهای واقع در بخشهای تکرشتهای و دست نخورده ماندن بخش دورشتهای، تغییر یافتن ناقص مولکولهای تحت تاثیر قرار گرفته، اتصال دوباره مولکولهای DNA تحت تاثیر غلظت بالای نمک و دسولفوناسیون ناقص میباشد. کیتهای تجاری بهینهشده میتوانند احتمال وقوع این موارد را کاهش دهند.