التهاب سلولهای میکروگلیال را به آزادسازی پروتئین ASC تحریک میکند. تحقیقات نشان دادهاست که ذرات ASC باعث تجمع و رسوب پروتئینهای بتا آمیلوئید در مغز موشها میشود. این علامت اصلی بیماری آلزایمر است.
از یک جهت بیماری آلزایمر نسبتا ساده به نظر میرسد: تودههای پروتئینی بتا آمیلوئید تشکیل میشوند و در نتیجه تجمع آنها، فرایندهای رو به زوالی رخ میدهند که در نهایت منجر به دمانس میشود. اما اگر همه عوامل موثر در وقوع بیماری را کنار همدیگر قرار دهیم، این جاست که متوجه میشویم آنقدرها هم ساده نیست و درواقع بسیار پیچیده است. به عنوان مثال نقش بالقوه فرایندهای التهابی در وقوع بیماری را درنظربگیرید. شواهد بسیاری مبنی بر وقوع التهاب در بیماری آلزایمر وجود دارد: سطح سلولهای ایمنی بالا میرود؛ سلولهای نوروگلیا واکنش شدیدی به محیط غیرطبیعی بافت میدهند؛ و تغییراتی در عروق سد خونی مغزی ایجاد میشود. علاوه بر اینها تغییرات ژنتیکی واضح در سلولهای گلیا احتمالا خطر ابتلا به بیماری آلزایمر را افزایش میدهد. با این حال علیرغم همه این شواهد، جزئیات چگونگی تاثیر التهاب بر پیشرفت بیماری به خوبی شناخته نشدهاست.
مقاله مرتبط: بیماری آلزایمر با مغز چه میکند؟
بتا آمیلوئید مشتق از یک پروتئین بزرگتر به نام پروتئین پیشساز آمیلوئید (Amyloid Precursor Protein) است. این پروتئین به قطعات کوچکتر تقسیم میشود تا گروهی از پپتیدهای کوچکتر را ایجاد کند. جهش در پروتئین APP یا در آنزیمهای پردازنده آن، میتواند به طور مستقیم به بیماری آلزایمر منجر شود؛ درصورتیکه باعث تولید بتا آمیلوئیدهایی شود که مستعد تجمع به صورت تودههای آسیبرسان محلول هستند. این تودههای محلول سپس بیشتر تجمع یافته و در بافت مغز به صورت پلاکهای آمیلوئیدی (amyloid plaques) رسوب میکنند. با این حال مکانیسم دقیق تشکیل پلاکها ناشناخته است. تاکنون همه آزمایشات بالینی فاز ۳ که برای تعیین درمانهای آینده انجام گرفتهاند، با شکست مواجه شدهاند. بنابراین شناخت مکانیسمهای مولکولی درگیر در تشکیل پلاک میتواند توسعه روشهای درمانی موثرتر را تسریع کند.
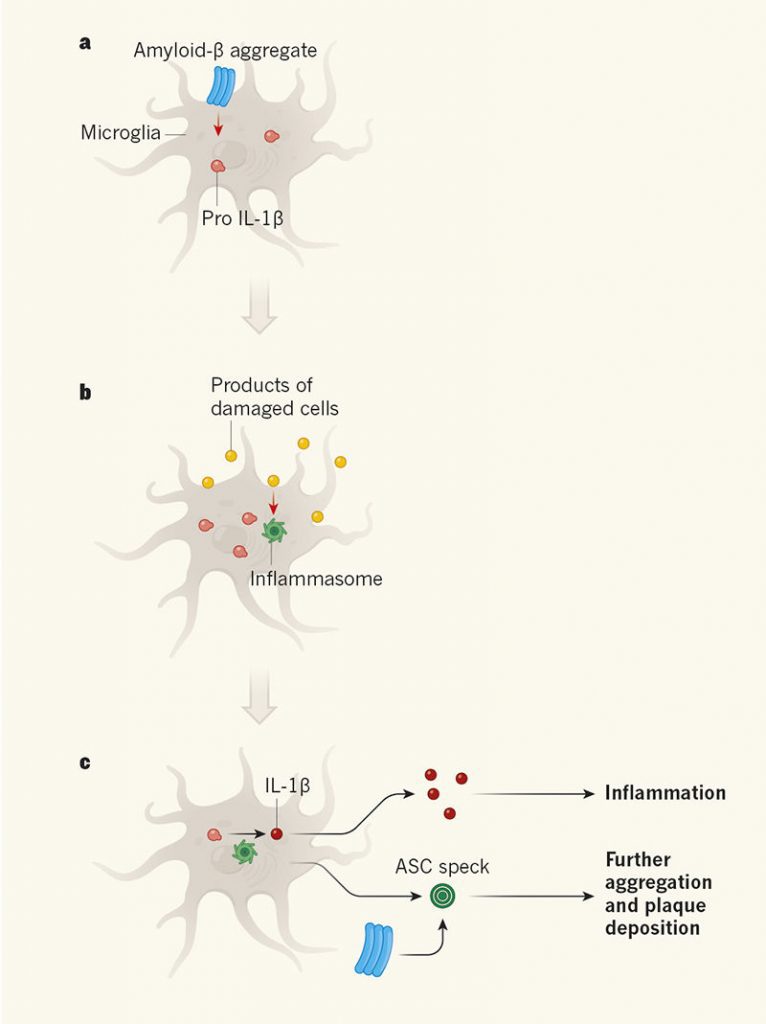
در این مطالعه Venegas و همکارانش از مشاهده پیشین خود که موشهای فاقد پروتئین NLRP3 نوعی مقاومت دربرابر رسوب بتا آمیلوئید نشان میدهند، شروع کردند. این یافته از حذف ژن Nlrp3 در نژادی از موشها به نام APP/PS1 به دست آمد. این موشها اختصاصا به گونهای تغییر داده شدهاند تا ژنهای انسانی دارای جهشهای مرتبط با بیماری آلزایمر را بیان کنند. در نتیجه بیان این ژنها، در مغز موشها پلاکهای بتا آمیلوئید رسوب کرده و اختلالات شناختی پیدا میکنند. NLRP3 نوعی حسگر ایمنی است که در مغز، در سلولهای میکروگلیا یافت میشود که سلولهای مرتبط با ایمنی سیستم عصبی مرکزی هستند. این حسگر، مواد شیمیایی غیرعادی مانند محصولات سلولهای آسیب دیده را شناسایی کرده و شروع به تشکیل پروتئین درون سلولی بزرگی به نام inflammasome می کند. این پروتئین حاوی خود NLRP3، رشتههای دراز پروتئین ASC، و آنزیمی به نام caspase-1 میباشد.
شناختهشدهترین اثر inflammasome این است که باعث آزادسازی پروتئینهای پیشالتهابی اینترلوکین β-۱ (IL-1β) و IL-18 از سلولها میشود. این پروتئینها در پاسخ به سیگنالهای التهابی اولیه تولید میشوند که در بیماری آلزایمر، شکل محلول پروتئین بتا آمیلوئید است. IL-1β و IL-18 از واسطههای ایمنی قدرتمندی هستند که نقش مهمی در فرایندهای دفاعی و پاسخهای التهابی دارند. احتمالا بهعلت این اثرات قدرتمند است که اینترلوکینها تا قبل از تشکیل inflammasomeها در داخل سلول باقی میمانند تا اطمینان حاصل شود که فقط در مواقع لزوم آزاد میشوند.
به دنبال تشکیل inflammasome، ذرات پروتئین ASC (ASC specks)، از این سلولها آزاد میشوند. در بسیاری از سلولهای غیرایمنی، آزاد شدن ASC با مرگ سلولی همراه است. اما سلولهای ایمنی از جمله میکروگلیاها، بهتر میتوانند آزاد شدن ASC را تحمل کنند. ASC در این سلولها باعث گسترش التهاب میشود؛ از آن جهت که ذرات ASC وارد میکروگلیاهای اطراف شده و در آنها نیز آزادسازی اینترلوکین اضافی را تحریک میکنند.
Venegas و همکارانش دریافتند که ممکن است علت مقاومت نسبت به رسوب بتا آمیلوئید در موشهای فاقد NLRP3، مربوط به ذرات پروتئینی ASC باشد. محققان ابتدا در محیط in vitro، پروتئینهای ASC را با بتا آمیلوئید مخلوط کردند. آنان مشاهده کردند که تودههای مشابه آمیلوئید در تراکم بالا به سرعت در حضور ASC شکل گرفتند و در ساختار این تودهها خود ASC نیز مشاهده شد.
در آزمایشات in vivo، Venegas و همکارانش مشاهده کردند که موشهای APP/PS1 که فاقد ASC بودند، رسوب بتا آمیلوئید کمتری در مغز داشتند و در تستهای حافظه فضایی عملکرد بهتری نسبت به موشهایی که ASCدر آنها وجود داشت، نشان دادند. در قدم بعدی، محققان عصاره مغزی موشهای پیر APP/PS1 را به موشهای جوان از همان نوع تزریق کردند. عصاره مغزی میتواند رسوب بتا آمیلوئید را در مغز موشهای جوان APP/PS1 تحریک کند؛ اما نه در صورتیکه این موشها به گونهای تغییر یابند که فاقد ASC شوند. به طرز مشابهی، عصاره مغزی موشهای پیر APP/PS1 فاقد ASC نیز توانایی ایجاد پلاک آمیلوئیدی کمتری را در مغز موشهای جوان از همان نوع داشت.
درنهایت محققان دریافتند که آنتیبادیهای ASC، از اتصال ذرات این پروتئین به سایر پروتئینها جلوگیری کرده و در نتیجه مانع از تشکیل تودههای مشابه آمیلوئید در شرایط ترکیب ذرات ASC با بتا آمیلوئید در شرایط in vitro میشوند. در شرایط in vivo، مشاهده شد که در صورت تزریق همزمان عصاره مغزی موشهای APP/PS1 و آنتیبادیهای ASC، رسوب بتا آمیلوئید در مقایسه با شرایط عدم تزریق آنتیبادی، کاهش مییابد.
مقاله مرتبط: آستروسیتها در بروز بیماری آلزایمر نقش دارند!
شواهدی که توسط Venegas و همکارانش به دست آمدند در مجموع نشان میدهند ذرات ASC رسوب بتا آمیلوئید را در موشهای APP/PS1 افزایش میدهند. آزمایشاتی که گروه انجام داد بسیار چالشبرانگیز بودند؛ چون هر دو نوع پروتئین ASC و بتا آمیلوئید هم با همدیگر تجمع پیدا میکنند و هم با سایر پروتئینها. با این حال آنان با تعیین کنترلهای مناسب توانستند ثابت کنند که پدیده مشاهده شده حاصل فعل و انفعالات بیولوژیکی معنی دار است و یک تجمع تصادفی و نامشخص نیست.
آیا این مطالعه جهت روشهای درمانی آینده را مشخص میکند؟ در حال حاضر، ارتباطات محکمی بین ژنهای تاثیرگذار بر ابتلا به بیماری آلزایمر و مسیر NLRP3-inflammasome-ASC شناخته نشده است و اگر شناخته شود میتواند به تعیین مسیر آینده کمک کند. در موشها به نظر میرسد که عملکرد inflammasome روند پیری مغز را با ایجاد مقادیر پایینی از التهاب، سرعت میبخشد. این التهاب هم سیستمیک است و هم در سیستم عصبی مرکزی رخ میدهد و و با سن و چاقی در دوران میانسالی در ارتباط میباشد. هر دو عامل ریسک فاکتورهای بیماری آلزایمر محسوب میشوند.
برای موثرتر کردن درمانهای عمومی بیماری آلزایمر باید بدانیم که کدام مسیرها باید هدف قرار داده شوند. مهار مسیرهای کلیدی و ابتدایی مانند مسیر تشکیل inflammasome با واسطه NLRP3، میتواند اثرات بهتری داشته باشد. اما این احتمال که این نوع درمانها با سیستم دفاعی بیمار سازگار شوند نیز وجود دارد. در مقابل هدف قرار دادن مسیرهای فرعی تر مانند جلوگیری از تشکیل ذرات ASC در مغز، اختصاصیتر است اما ممکن است نتواند به طور کارآمدی مکانیسمهای فراوان درگیر در بیماری آلزایمر را مهار کند. با وجود این مشکل، کشف یک مکانیسم که میتواند به عنوان یک هدف درمانی مورد استفاده قرار گیرد بسیار دلگرم کننده است.