SCID طبقه بندی و تظاهرات بالینی
SCID شامل تعدادی از اختلالات نادر و مونوژنیک است که ویژگی مشترک آنها، وقوع یک مانع در تمایز سلولهای T با اختلال مستقیم یا غیر مستقیم در ایمنی سلولهای B است. شیوع کلی SCID بین ۱:۵۰۰۰۰ یا ۱:۱۰۰۰۰۰ تولد تخمین زده شده است. که احتمالا مرگهای زود هنگام از تشخیص صحیح جلوگیری کرده و شیوع کمی کمتر محاسبه شده است. مطالعه الگوی وراثت، ویژگیهای ایمونولوژیک و ژنوتیپ حداقل ۱۱مدل متفاوت از SCID را مشخص میکند.
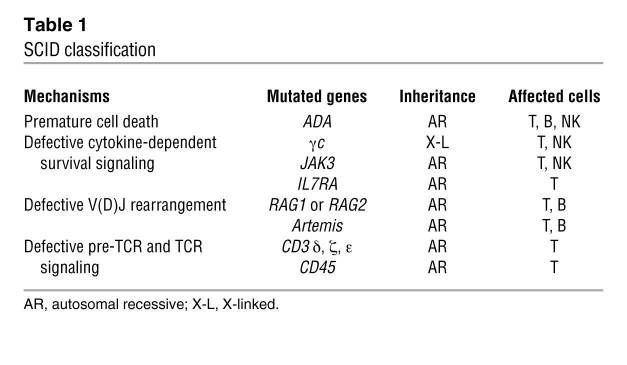
چهار مکانیسم اصلی که برای این بیماری تعریف شده است شامل:
۱. مرگ سلولهای نابالغ در اثر افزایش متابولیسم پورین، همانطور که در نقص آدنوزین دآمیناز (ADA) مشاهده شد.
۲.سیگنالهای بقای وابسته به سایتوکاین معیوب در پیش سازهای T cell (و بعضا پیشسازهای NK cellها) ، که این مکانیسم بیش از نیمی از بیماریهای SCID را به خود اختصاص میدهد. نقص در بیان یا عملکرد زیرواحد رسپتور سایتوکاین γ مشترک (γc) که مشترک با گیرندههای IL-2, IL-4, IL-7, IL-9, IL-15 و IL-21 است باعث فرم وابسته به X این بیماری میشود. (SCID-X1) و با کمبود کامل لنفوسیتهای T و NK همراه است. نقص در JAK3 با منطقه سیتوپلاسمی γc همراه است، موجب یک فنوتیپ مشابه میشود.
۳.نقص بازآرایی V(D)J برای ژنهای رسپتور TCR و B cell .در آزمایش ما این گروه حدود ۳۰% موارد SCID را شامل میشوند. نقص در هر کدام از RAG1 یا RAG2 (نوترکیبی لنفوئیدی مخصوص عناصر آغازگر) و یا Artemis (عامل درگیر در اتصال پایانی مسیر بازسازی غیرهمولوگ) منجر به نقص بازآرایی V(D)J و در نتیجه مرگ تیموسی و B-cell نابالغ میشوند.
۴.نقص pre-TCR و سیگنالینگ TCR. نقص کامل T-cell در اثر نقص پیش ساز CD3 (مثل: CD3δ, CD3ε, or CD3ζ) و یا پروتئینهای اصلی CD45 تیروزین فسفاتاز که در مرحله انتخاب مثبت pre-TCR و یا TCR سیگنالینگ اند.
برخی محققان نقصهای دیگر T cell ها را نیز جزو گروه SCID در نظر میگیرند. مثل: نقص ZAP-70 ، نقص CD3γ ،نقص بیان HLA class II، نقص پورین نوکلئوزید فسفوریلاز ، نقص لیگاز IV و یا Cernunnos و همچنین سندروم Omenn. با این حال، از آنجاییکه این شرایط با حضور T-cellهای بالغ (هرچند نقص عملکردی داشته باشند)، مسائل متفاوت ( در پایین آورده شده است) از نگرانیهای درمان را مطرح میکنند، در این نوشته به آنها نپرداختهایم.
تظاهرات بالینی SCID های مختلف نسبتا یکنواخت است و با عفونت زود هنگام (معمولا در دستگاه تنفسی و روده) مشخص میشود. ارگانیسمهای فرصت طلب مشترک مانند Pneumocystis cariniiو Aspergillus و ارگانیسمهای داخل سلولی مثل سایتومگالوویروس میتوانند عفونتهای مکرر و نارسایی رو به رشد را باعث شوند. شدت این تظاهرات بالینی SCID را به یک بیماری اورژانسی تبدیل میکند، که در غیاب درمان منجر به مرگ در سال اول زندگی میشود.
پیوند آلوژنیک HSC یا همان HSCT برای درمان نقص ایمنی اولیه: موفقیتها ومحدودیتها
با شروع این تجربه پیشگام در سال ۱۹۸۶، در سراسر جهان صدها بیمار SCID و صدها بیماری که مبتلا به نقص ایمنی اولیه کشنده بودند از HSCT بهره بردند. در حال حاضر، HSCT اهدایی از خواهر یا برادری که HLA هماهنگ دارند، حداقل ۸۰% شانس درمان برای کودکان مبتلا به نقص ایمنی اولیه ایجاد میکند و حدود ۷۰% شانس درمان برای HLA هماهنگ اما غیر فامیل وجود دارد. این درصد موفقیت بالا درنتیجهی مدیریت بهتر مسائل تغذیهای و مشکلات عفونتی است که این بیماران در زمان درمان با آن مواجه میشوند.
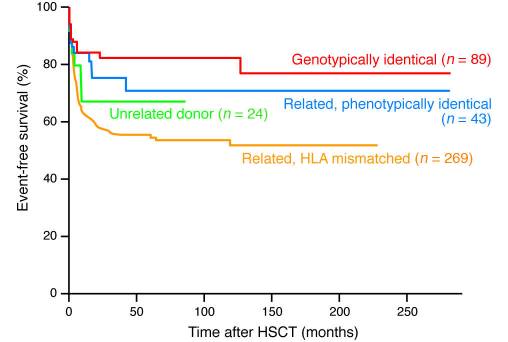
برعکس زمانیکه اهدا از وابستگان اما با HLA غیرهماهنگ صورت بگیرد ( مثلا زمانیکه یکی از والدین به فرزند اهدا میکند) میزان بقا به طور قابل توجهی پایینتر از دو مورد قبل است و این علیرغم وجود روشهای ex vivo مختلف برای از بین بردن T-cell های بالغ اهدایی (مسئول وقوع شدید واکنش پیوند در میزبان) در مغز استخوان گرفته شده است.
گستردگی استفاده از اهدا از بستگان با HLA ناهماهنگ محدود به موارد زیر است:
۱.سن دریافت کننده در زمان پیوند (مرگ ومیر در کودکان بزرگتر بیشتر است که با بیمارهای عفونی در ارتباط است)
۲.عوارض ایمونولوژیک (مانند واکنش پیوند در میزبان) به دلیل اختلاف زیاد HLA در دهنده و گیرنده.
۳.بازسازی ایمنی آهسته و حتی جزئی، مسئول عوارض دیرهنگام شامل عفونت، التهاب و حوادث خودایمنی
۴.کاهش طولانی مدت عملکرد سلولهای T ، مربوط به نبود سلول بنیادی اهداکننده پیوند و احتمالا کاهش قبل از بلوغ در عملکرد تیموس، زمانیکه به بیمار SCID دیر پیوند زده شود.
در آینده، با پیشرفت در زمینه کنترل واکنش آلوژنیک و بالا رفتن سرعت بازسازی ایمنی انتظار میرود این موانع از بین برود. و از عملکرد HSCT وابستگان با HLA ناهماهنگ در بیماریهایی غیر از نقص ایمنی (مانند هموگلوبینوپاتی و اختلالات بدخیمی) جلوگیری میشود. در عین حال، یک فرایند ژن درمانی (مبتنی بر HSCT اتولوگ ژن اصلاح شده) میتواند محدودیتهای ذکر شده را دور زده و یک جایگزین درمانی مناسب به نمایش بگذارد.