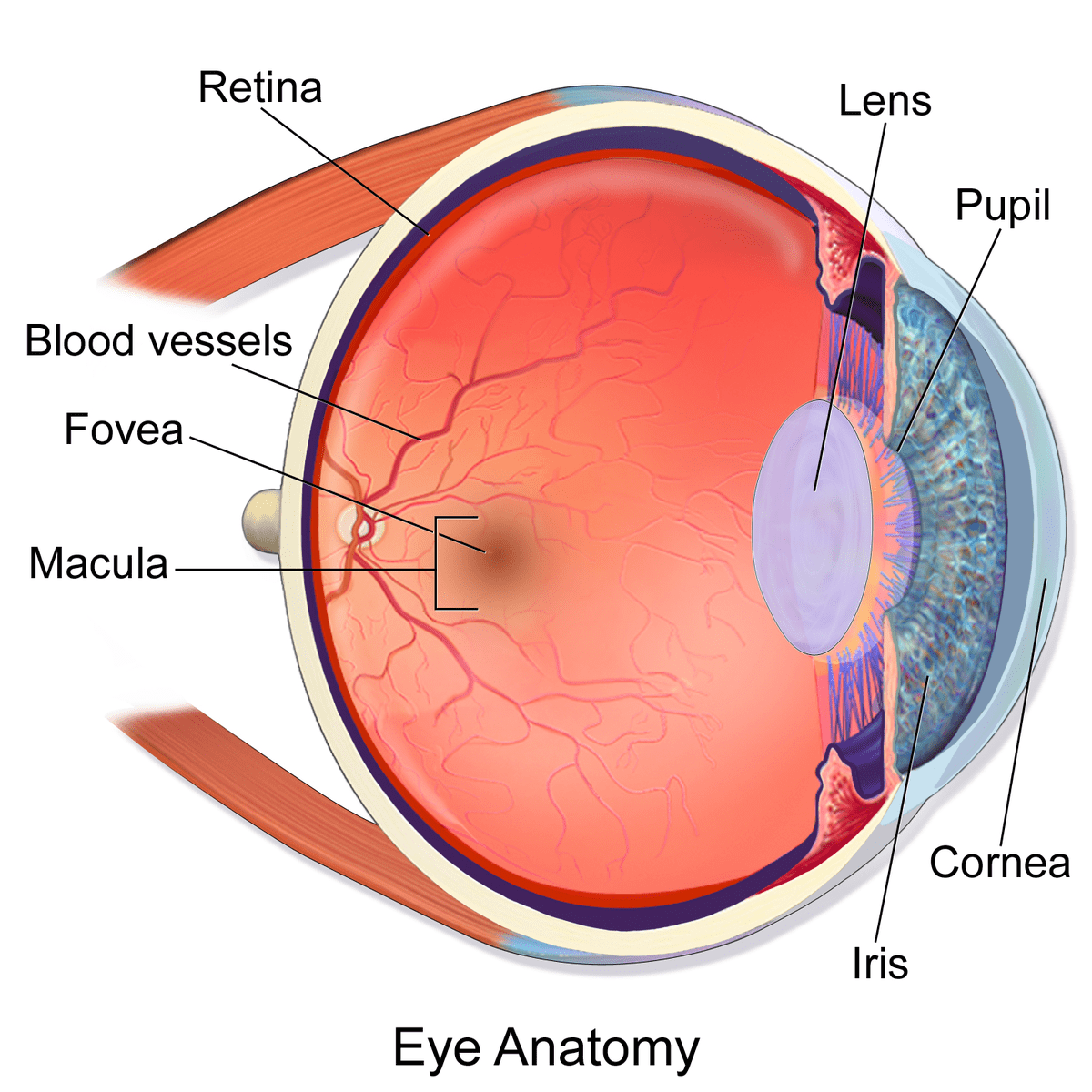
از نظر بسیاری از افراد، بینایی مهمترین حس در میان حواس پنجگانه است درک بینایی به واسطهی شبکیه انجام میگیرد، بافت حساس به نوری که لایه داخلی چشم را میپوشاند. نوری که به شبکیه برخورد میکند آبشاری از رخدادها را آغاز میکند و در نهایت ایمپالسهای عصبی به سمت مغز فرستاده میشود. شبکیه یک ساختاری پیچیده و لایه لایه است که از سلولهای عصبی و پشتیبان تشکیل شده است. فتورسپتورها سلولهای حساس به نوری هستند که نور را از میدان دید دریافت کرده و این داده از محیط بیرون را به تغییر پتانسیل غشا (پیام عصبی) تبدیل میکند. دو نوع فتورسپتور در چشم وجو دارد: گیرندههای مخروطی و استوانهای. سلولهای استوانهای به دریافت نور در تاریکی کمک کرد و سلولهای مخروطی امواج نور در روشنایی را دریافت کرده و موجب درک رنگها و جزئیات ظریف اشیاء میشوند. گیرندههای مخروطی در بیشتر در قسمت مرکزی شبکیه تجمع دارند و این ناحیه تحت عنوان لکه زرد شناخته میشود، و بیشترین تراکم این گیرندهها در ناحیهی گودی مرکزیِ لکه زرد (fovea) است، این ناحیه مشاهدهی جزئیات ظریف اشیاء را ممکن میکند. لایه رنگدانهای شبکیه (RPE) سلولهای گیرنده نور را پشتیبانی و تغذیه میکند و به طور کاملاً محکم به لایهی مشیمیه که حاوی مویرگهای فراوان است، متصل شده است. لایه رنگدانهای شبکیه چرخه بینایی را میانجیگری میکند، یک پروسه مداوم که در آن ترکیبات شمیایی به نام رتینوئیدها، که در تبدیل اطلاعات میدان دید به پیام عصبی مورد استفاده قرار میگیرند، بازیابی میشوند.
دیستروفی شبکیه: ویژگی بارز دیستروفی شبکیه دژنره شدن فتورسپتورها و سلولهای لایه رنگدانهای شبکیه است. بیماریهای ناشی از دیستروفی شبیکیه دلیل عمدهی نابینایی ارثی و غیرقابل درمان هستند. به طور کلی این بیماریها بر اساس نوع آسیب (آسیب بر گیرندههای مخروطی، استوانهای، آسیب لکه زرد و یا نواحی محیطی شبکیه) طبقهبندی میشوند. تفاوت بیین بیماریهای مختلف دیستروفی شبکیه ممکن است بسیار ناچیز باشد زیرا جهش در یک ژن منفرد منجر به علایم بالینی متنوعی میشود.
نابینایی ارثی ممکن است ناشی از نقص ژنی در سلولهای لایه رنگدانهای شبکیه باشد -جایی که تداوم چرخه بینایی پشتیبانی میشود- و یا ممکن است به علت نقص ژنی در فتورسپتورها بوجود آید. یکی از اجزای کلیدی چرخه بینایی رتینويد ایزومرازها هستند که از طریق ژن RPE65 کد میشوند و موتاسیون در این ژن سبب بیماری لبرآموروزیس مادرزادی (LCA) میشود.
لبر آموروزیس مادرزادی (Leber Congenital Amaurosis)
بیماری لبر درجات مختلفی از اختلال های رنگدانه، نازک شدن عروق و از بین رفتن عصب بینایی را به همراه دارد. این اختلال معمولاً در زمان تولد یا اندکی پس از آن بروز کرده و عامل ایجاد اختلال بینایی و یا نابینایی در کودکان است. لبر تخریب یا از بین رفتن پیش از تولد شبکیه است. به نظر میرسد حدود ۱۰ تا ۲۰ درصد کودکان نابینا از لبر رنج میبرند واین موضوع لبررا یکی از متداولترین علل نابینایی کودکان قرار میدهد. بیماران لرزش چشم و اغلب چشمهای گود رفته دارند. حساسیت شدید نسبت به روشنایی نور همراه با این بیماری اتفاق میافتد و نیز تعدادی از بیماران مبتلا به LCA ناهنجاریهای سیستم اعصاب مرکزی را نشان میدهند.
علائم بالینی: افراد مبتلا به لبر از زمان تولد افت شدید بینایی دارند. ظرف مدت کوتاهی در نخستین ماههای زندگی کودک، اغلب والدین متوجه نقص در واکنشهای بینایی و حرکات سر گردان و غیر طبیعی چشم (نیستاگموس) میشوند. در این زمان، معاینات چشمی کودکان مبتلا به لبر، تظاهرات طبیعی را در شبکیه نشان میدهد؛ با این حال، اندازهگیری میزان عملکرد بینایی توسط ERG فعالیت اندک شبکیه را نشان میدهد. بنابراین آزمایشات ERG عامل اصلی تشخیص لبر می باشند. در ابتدای جوانی، بلافاصله تغییرات گوناگون در شبکیهی بیماران لبر بروز میکند. عروق خونی اغلب باریک وتنگ میشود. دامنهای از تغییرات لایه رنگدانهای شبکیه اتفاق میافتد. گاهی اوقات تغییرات رنگدانهای ،شبیه به دیگر بیماری های دیستروفی شبکیه مثل رتینت پیگمانتوزا (RP) هستند. در هرحال با افزایش سن تغییرات بارزی در شبکیه ایجاد میشود، اغلب به ندرت بینایی تا دوران جوانی ثابت میماند وگذشت زمان پیش آگهی بینایی باقیمانده را مشخص میکند. قدرت بینایی در بیماران لبر، اغلب محدود به شمارش انگشتان یاحرکت دست یا تشخیص روشنایی نور است. بعضی بیماران کاملاً نسبت به نور گریزان بوده و ترس از نور دارند. بیمارانی که باقیماندهای از بینایی دارند، اغلب بسیار دوربین یا نزدیکبین هستند. بسیاری از کودکان مبتلا به لبر، عادت فشار دادن چشمها از لحاظ بالینی، به عنوان رفلکس چشمی _ انگشتی شناخته میشود. چشمان افراد مبتلا به لبر اغلب ظاهر گود رفته یا فرورفته دارند. همچنین قوز قرنیه و آب مروارید همراه با این بیماری گزارش شده است. در تعدادی از نمونهها، لبر همراه با بعضی عوارض سیستم مرکزی، مثل تاخیر رشد، صرع و نقص مهارتهای حرکتی میباشد، وابستگی اندک لبر با تعدادی عوارض سیستم اعصاب مرکزی شناخته شده است.
ویژگی بارز بیماری لبر از بین رفتن شدید بینایی در مراحل اولیه زندگی و تخریب پیشروندهی سلولهای شبکیه است. از نظر ژنتیکی، تقریباً تمامی انواع بیماریهای LCA اتوزومال غالب هستند و شیوع آن در سراسر جهان یک نفر در هر ۳۰۰۰۰ نفر است. تا کنون موتاسیون در ۱۸ ژن مختلف برای این بیماری گزارش شده است. موتاسیون در ژن RPE65 ۱۰ درصد از کل موارد را شامل میشود و تحت عنوان LCA2 شناخته میشود. در ژندرمانی، جایگزینی ژن RPE65 بوسیلهی وکتورهای AAV، به عنوان یک استراتژی درمانی موفق برای LCA2 به کار گرفته میشود.
| Type | OMIM | Gene | Locus |
| LCA1 | ۲۰۴۰۰۰ | GUCY2D,[۱۰] | ۱۷p13.۱ |
| LCA2 | ۲۰۴۱۰۰ | RPE65[۱۱] | ۱p31.3-p31.۲ |
| LCA3 | ۶۰۹۸۶۸ | SPATA7 | ۱۴q31.۳ |
| LCA4 | ۶۰۴۳۹۳ | AIPL1[۱۲][۱۳] | ۱۷p13.۲ |
| LCA5 | ۶۰۴۵۳۷ | LCA5[۱۴] | ۶q14.۱ |
| LCA6 | ۶۰۵۴۴۶ | RPGRIP1 | ۱۴q11.۲ |
| LCA7 | ۶۰۲۲۲۵ | CRX[۱۵] | ۱۹q13.۳ |
| LCA8 | ۶۰۴۲۱۰ | CRB1 | ۱q31-q32.۱ |
| LCA9 | ۶۰۸۵۵۳ | NMNAT1[۱۶][۱۷][۱۸][۱۹] | ۱p36.۲۲ |
| LCA10 | ۶۱۰۱۴۲ | CEP290 | ۱۲q21.۳۲ |
| LCA11 | ۱۴۶۶۹۰ | IMPDH1 | ۷q32.۱ |
| LCA12 | ۱۸۰۰۴۰ | RD3 | ۷q32.۱ |
| LCA13 | ۶۰۸۸۳۰ | RDH12 | ۱q32.۳ |
| LCA14 | ۶۰۴۸۶۳ | LRAT | ۱۴q24.۱ |
| LCA15 | ۶۰۲۲۸۰ | TULP1 | ۴q31 |
| LCA16 | ۶۰۳۲۰۸ | KCNJ13 | ۲q37 |
| LCA17 | ۶۰۱۱۴۷ | GDF6 | ۸q22 |
| LCA18 | ۱۷۹۶۰۵ | PRPH2 | ۶p21 |
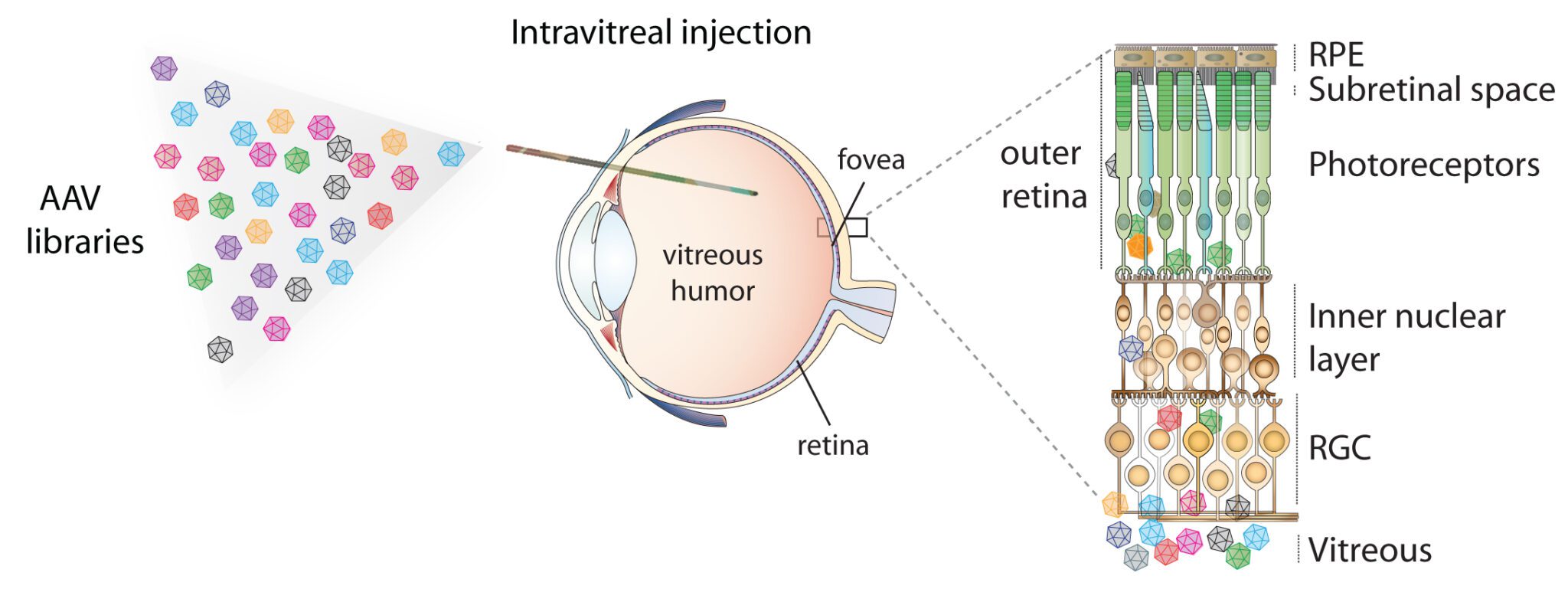
همانطور که توضیح داده شد برای درمان بیماری لبر قطعه سالم ژن RPE65، درون وکتورهای AAV، به چشم بیمار تزریق میشود. این تزریق به صورت تحت شبکهای (subretinal) انجام میگیرد (درصورتیکه آتروفی لکه زرد در OCT مشاهده نشود، تزریق در این ناحیه لکه زرد صورت نمیگیرد تا از عوارض جانبی مانند جدا شدن ماکولا و لکه زرد جلوگیری شود). میتوان با تزریق به سه ناحیهی شبکیه فوقانی (در نواحی اطراف ماکولا و خارج از لکه زرد)، شبکیه تمپورال و شبکیه نازال فوقانی، نواحی آلوده شده با وکتور را افزایش داد و اثرگذاری درمان را بالا برد.
همچنین در کارآزماییهای بالینی انجام شده، طی آزمایش PCR انجام شده پس از تزریق، در خون محیطی بیماران هیچگونه ژنومی از وکتور مشاهده نشد؛ بنابریان وکتور AAV به طور کاملاً اختصاصی فقط روی شبکیه اثر میگذارد. در کارآزماییهای بالینی مختلف پاسخ ایمنی هومورال نیز از طریق سنجش میزان آنتیبادی تولید شده علیه کپسید AAV بررسی شد و پس از تزریق افزایش تیتر آنتیبادی در سرم مشهود نبود.
 با توجه به افزایش چشمگیر حدت بینایی (VA)، کاهش نیستاگموس، عدم وجود عوارض جانبی (انتشار سیتماتیک وکتور یا پاسخ ایمنی خطرناک علیه وکتور)، این درمان که با نام لوکستورنا (Luxturna) شناخته میشود، مورد تایید FDA قرار گرفت. لوکستورنا از طریق انتقال ۱۵۰ بیلیون وکتور، که حامل کپی سالمی از ژن RPE65 هستند، عمل میکند و فقط برای یک بار تزریق طراحی شده است.
با توجه به افزایش چشمگیر حدت بینایی (VA)، کاهش نیستاگموس، عدم وجود عوارض جانبی (انتشار سیتماتیک وکتور یا پاسخ ایمنی خطرناک علیه وکتور)، این درمان که با نام لوکستورنا (Luxturna) شناخته میشود، مورد تایید FDA قرار گرفت. لوکستورنا از طریق انتقال ۱۵۰ بیلیون وکتور، که حامل کپی سالمی از ژن RPE65 هستند، عمل میکند و فقط برای یک بار تزریق طراحی شده است.
با توجه به اینکه لبر یک بیماری پیشرونده است، سوالی که مطرح میشود این است که آیا سن بیمار در روند درمان تاثیرگذار است یا نه؟ درواقع فنوتیپ لبر مرتبط با ژن RPE65، نه تنها شامل عدم کاکرد شدید بینایی است بلکه تخریب پیشروندهی شبکیه را نیز در پی دارد. ارتباط بین شدت تخریب شبکیه و سن پیچیده است. در یک بیمار منفرد به طور قطع شدت بیماری با افزایش سن، به خصوص در ۳ دههی اول زندگی، افزایش مییابد اما روند افزایش شدت بیماری در تمامی بیماران یکسان نیست. در برخی بیماران شدت تخریب و از بین رفتن بینایی در دهه اول و در برخی دیگر از بیماران در دهه سوم زندگی افزایش مییابد. با محاسبه تعداد فتورسپتورهای باقیمانده در شبکیه و تزریق وکتورها به ناحیه مناسب، همهی بیماران شانس برابری در دریافت درمان مناسب خواهند داشت؛ بنابراین اثرگذاری درمان مستقل از سن بیمار است.
با وجود اینکه این شیوه درمانی یک دستآورد علمی مهم است اما هزینهی درمانی بالای آن مانعی برای دسترسی همگان است. لوکستورنا توسط شرکت داروسازی Spark طراحی شده است و قیمت آن برای هر درمان، ۸۵۰ هزار دلار در نظر گرفته شده است.
سلام من به مدت بیست سال است که دچار بیماری ارپی شده ام می خواستم سوال کنم درمان این بیماری که درمورد ان توضیح داده شد تا چه زمانی در ایران انجام می شود با تشکر