تکنیک inverse PCR (IPCR) در سال ۱۹۸۸ توسط Ochmen و همکارانش ایجاد شده و نخستین استراتژی انجام فرایند genome walking میباشد. اساس این تکنیک PCR استاندارد است. inverse PCR روشی برای تکثیر DNAای با توالی ناشناخته (flanking region) است که بلافاصله در مجاورت ناحیهای با توالی معین (core region) قرار دارد و برای تکثیر آن طراحی پرایمر اختصاصی ممکن نیست. این توالی میتواند یک ترنسپوزون و یا یک انکوژن با توالی شناخته شده باشد.
در فرایند PCR عادی از پرایمرهای الیگونوکلئوتیدی استفاده میشود که به رشتههای مخالف هم متصل میشوند و جهت قرارگیری آنها به گونهای است که توالی بین دو پرایمر تکثیر میگردد. در این حالت محصولات تولیدشده به عنوان توالی الگو مورداستفاده قرار میگیرند و درنتیجه واکنش تکثیر به صورت نمایی انجام میشود. با این حال در این فرایند، توالیهایی که بلافاصله خارج پرایمر قرار دارند، غیرقابل دسترسی هستند؛ چون پرایمرهایی که باعث تکثیر DNA به سمت نواحی اطراف (به جای توالی مرکزی) میشوند، تنها امکان افزایش خطی تعداد کپیها را فراهم میکنند. علت این موضوع نبود پرایمر reverse ای است که در جهت عکس و از رشته مخالف عمل تکثیر را انجام دهد.
مقاله مرتبط: PCR چیست؟
inverse PCR تکنیکی است که از اصول PCR عادی استفاده میکند و برخلاف آن دارای قابلیت تکثیر توالیهای موجود در هر دو سمت توالی مرکزی است و از این رو کاربردهای فراوانی در ژنتیک دارد. با انجام Inverse PCR میتوان توالیهای ژنومی را بازیابی و به منظور توالییابی استفاده کرد. جهت قرارگیری پرایمرها در inverse PCR عکس PCR عادی است و درنتیجه تکثیر نیز در جهت عکس حالت استاندارد انجام میگیرد. از این رو است که این تکنیک inverse PCR یا PCR معکوس نام گرفته است.
از جمله این کاربردهای این تکنیک میتوان به تعیین توالیهای مربوط به الحاق ترانسپوزون و توالییابی اشاره کرد. همانطور که گفته شد، inverse PCR میتواند برای تکثیر سریع و موثر قطعاتی از DNA با توالی ناشناخته که دو طرف هر گونه توالی شناخته شده در DNA ژنومی یا cDNA را احاطه کردهاند، به کار رود. این تکنیک نیاز به ساخت کتابخانه DNA و یا بررسی آن به منظور شناسایی توالیهای ناشناخته را از بین میبرد. در ساخت کتابخانه DNA مشکل این است که برخی از فاژها یا پلاسمیدهای نوترکیب در باکتریها ناپایدار هستند. inverse PCR این مشکل را رفع میکند.
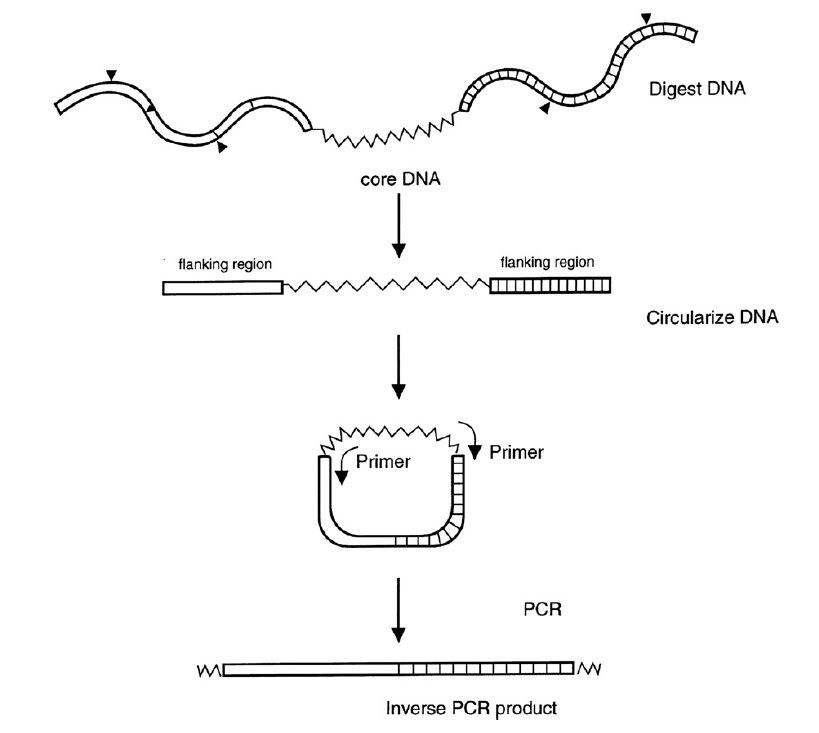
Invese PCR دارای سه مرحله است (تصویر ۲):
- تجزیه DNA الگو توسط آنزیم محدود کننده
- حلقوی کردن DNA تجزیه شده
- فرایند تکثیر توسط PCR
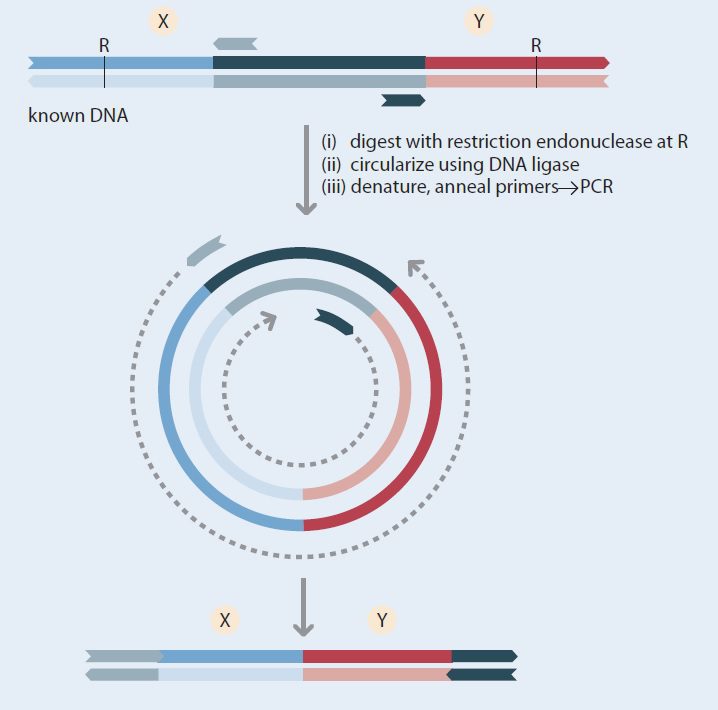
طی نخستین مرحله، DNA ژنومی توسط آنزیم محدودکننده تجزیه میشود. آنزیم مورد استفاده نباید هیچ نقطه برشی درون توالی شناخته شده داشته باشد و برش باید در دو طرف این توالی انجام گیرد؛ به گونهای که طول قطعات حاوی توالی موردنظر در حدود ۳-۴ کیلوباز باشد. در صورتیکه آنزیم به گونهای انتخاب شود که در داخل توالی مرکزی نیز نقطه برش داشته باشد، محصولات تولید شده طی inverse PCR تنها دارای یکی از دو توالی اطراف خواهند بود.
تعداد نقاط برش آنزیم در DNA الگو یک فاکتور تعیینکننده است. به عنوان مثال، اگر تعداد این نقاط اندک باشد، اندازه قطعات تشکیلشده به طور میانگین بزرگ خواهد بود و در این صورت تکثیر توسط PCR به طور موثر انجام نخواهد شد. برای تعیین آنزیم محدودکننده مناسبی که بتواند قطعاتی با این مشخصات تولید کند، میتوان به صورت تجربی از تکنیک Southern-blot و یا هیبریداسیون پروب (به نحوی که کل توالی مرکزی یا بخشی از آن به پروب متصل شود)، استفاده کرد. در پایان این مرحله باید استخراج قطعات DNA تشکیل شده انجام گیرد تا مولکولهای DNA از آنزیم جداسازی شوند. همچنین برای رسیدن به این هدف میتوان مخلوط DNA را به مدت ۱۵-۲۰ دقیقه در دمای ۶۵ درجه سانتیگراد حرارت داد تا آنزیمهای محدودکننده غیرفعال شوند.
در دومین مرحله مخلوط DNA رقیقسازی شده و تحت تاثیر DNA لیگاز قرار میگیرد. علت رقیقسازی این است که بازده اتصالات درون مولکولی به حداکثر رسیده و احتمال تولید مولکولهای حلقوی مونومریک بالاتر رود. در صورت استفاده از غلظت بالای مخلوط DNA، سطح اتصالات هتروژنیک بالاتر رفته و میزان تکثیر غیراختصاصی افزایش مییابد.
T4 DNA Ligase میتواند به عنوان آنزیم استفاده شود. این فرایند نیاز به ATP دارد که باید به مخلوط واکنش افزوده شود. در نهایت انتهاهای مکمل هر مولکول DNA تشکیلشده که تعداد کل آنها در تجزیه DNA ژنومی ممکن است به هزاران عدد نیز برسد، به همدیگر متصل و مولکولهای DNA حلقوی تشکیل می شوند. این مولکولها هستند که به عنوان الگو در PCR استفاده خواهند شد.
پیش از انجام فرایند PCR دو نوع واکنش کنترل باید طراحی شوند: نخستین کنترل حاوی تمام مواد اولیه واکنش به جز DNA الگو است. در واکنش کنترل دوم، پلاسمیدی با سایز مشخص که DNA حاوی پرایمرهای الیگونوکلئوتیدی به آن الحاق شده است، به جای DNA الگو به کار میرود.
در سومین مرحله فرایند PCR انجام میگیرد. پرایمرهای واکنش PCR به گونهای طراحی میشوند که بتوانند به دو طرف توالی شناختهشده متصل شوند. اما جهت تکثیر آنها عکس یکدیگر، رو به خارج از این توالی و به سمت توالی ناشاخته مجاور است و به عبارت دیگر معکوس است. هر پرایمر باید طولی در حدود ۲۰-۳۰ نوکلئوتید داشته و حاوی مقادیر یکسانی از هر ۴ باز با توزیع متعادل بازهای G و C باشد. میتوان به انتهای ۵’ این پرایمرها restriction site اضافه کرد تا کلون کردن در مراحل بعدی تسهیل شود. نتیجه فرایند تکثیر تولید مولکولهای DNA خطی است که حاوی دو توالی ناشناخته موردنظر که در هر دو طرف ۳’ و ۵’ توالی مرکزی قرار داشتند، میباشد. اتصال این دو قطعه باعث تولید یک نقطه برش مخصوص آنزیم محدودکننده اولیه میشود.
مرحله دناتوراسیون در inverse PCR بسیار مهم است؛ چون مولکولهای حلقوی DNA اگر دو رشتهای بمانند، تمایل به تشکیل superhelix دارند و درنتیجه الگوی مناسبی برای PCR نیستند. در PCR دمای دقیق مرحله اتصال به طور تجربی و مطابق طبق خصوصیات جفت پرایمرهای مورد استفاده در واکنش تعیین میشود. پس از پایان فرایند PCR محصولات واکنشهای تست و کنترل باید توسط الکتروفورز در ژل آگارز یا پلیاکریلآمید بررسی شوند. محصولات ایجاد شده در یک واکنش PCR موفق باید به آسانی قابل مشاهده باشند. تعیین هویت نوار تشکیل شده میتواند توسط توالییابی، restriction mapping و Southern-blot انجام گیرد.
با سلام
ممنون از سایت بسیار خوبتون و نکات ارزشمندی که به اشتراک گذاشتید. یک سری ابهاماتی برای من پیش اومد ممنون میشم برطرف کنید.
من یک بخشی از invers PCR رو متوجه نشدم که فرموده بودید پرایمر reverse وجود نداره؛ پس چطوری سنتز رشته جدیدی از روی رشته مکمل الگو انجام میشه؟ سوال دیگری که پیش اومد اینه که محصول نهایی مثل PCR معمولی، خطی و دو رشته ای هست؟ اگر خطی هست چطوری این اتفاق میوفته؟!