سندرم هولت-اورام که تحت عنوان سندرم دست-قلب نیز شناخته میشود، نوعی ناهنجاری ارثی به صورت اختلال در اندامهای فوقانی و قلب است. هولت و اورام این بیماری را برای اولین بار در سال ۱۹۶۰ در چهار نسل از یک خانواده که همزمان به نفص سپتوم دهلیزی و ناهنجاری شست مبتلا بودند، توضیح دادند.
پاتوفیزیولوژی
این سندرم توارث اتوزومی غالب با الگوی کاملاً نافذ دارد. بیماری به علت جهشهایی در فاکتور رونویسی TBX5 است که این فاکتور در تکامل قلب و اندامهای فوقانی دخالت دارد. عوارض پاتوفیزیولوژیک نیز نتیجهی مستقیم مالفورماسیونهای قلب و اندامهای فوقانی است. هیچ فاکتور محیطی اثرگذاری نیز شناخته نشده است.
درگیری اندام فوقانی
با وجود متغیر بودن تظاهرات بالینی بیماری، اختلال و ناهنجاری اندامهای فوقانی همواره وجود دارد. این ناهنجاریها ممکن است یکطرفه یا دوطرفه و نامتقارن باشند. همچنین ممکن است استخوانهای رادیال، کارپال و تنار را درگیر کنند. آپلازی، هیپوپلازی، فیوژن یا آنومالی تکاملی این استخوانها، موجب طیفی از فنوتیپها شامل شست تریفالانژیال یا نبود شست میشوند. گاهی مالفورماسیون اندام فوقانی در حدی شدید است که باعث فوکوملیا (فوکوملیا نوعی مالفورماسیون است که در آن دستها به تنه چسبیدهاند.) میگردد؛ این شرایط سندرم تالیدومید کاذب نامیده میشود. شایعترین یافتهها در افراد مبتلا به سندرم هولت-اورام، مالفورماسیونها یا فیوژنهای استخوانهای کارپال است. ناهنجاری استخوانهای کارپال تنها یافتهای است که در همهی افراد مبتلا وجود دارد. هرچند این آنومالیها ممکن است در برخی بیماران تنها در رادیوگرافی مشهود باشند.
درگیری قلبی
تقریباً ۷۵ درصد از بیماران ناهنجاری قلبی دارند. در بیشتر بیماران، ناهنجاری به صورت نقص سپتوم دهلیزی (ASD) یا نقص سپتوم بطنی (VSD) با تعدد، سایز و موقعیت متغیر است. ASDها معمولاً از نوع سکوندوم هستند؛ در حالی که VSDها بیشتر در سپتوم ترابکوله عضلانی رخ میدهند. آنومالیهای قلبی در این بیماری همچنین شامل اختلالات هدایتی قلب همچون بلوک پیشروندهی دهلیزی-بطنی و فیبریلاسیون دهلیزی است. این آنومالیها غالباً حتی در غیاب نقایص سپتال نیز حضور دارند.
اپیدمیولوژی
ایالات متحده
سندرم هولت-اورام شایعترین حالت سندرم قلب-دست یا شیوع تخمینی ۰.۹۵ مورد در هر ۱۰۰ هزار تولد است که تقریباً ۸۵ درصد موارد در ارتباط با جهشهای جدید هستند.
مورتالیتی/موربیدیتی
پیشآگهی بیماری در حالت کلی خوب بوده؛ اما به شدت مالفورماسیونهای قلبی بستگی دارد. ضایعات ساختاری به هنگام تولد وجود داشته و پیشآگهی در واقع به شدت ضایعات قلبی وابسته است. چنانچه شانت داخل قلبی قابل توجه میتواند موجب مرگ ناگهانی یا بروز هایپرتنشن ریوی و سندرم آیزنمنگر شود. اولین تظاهر بالینی بیماری ممکن است نارسایی قلبی، آریتمیهای قلبی از جمله بلوک قلبی یا اندوکاردیت عفونی باشد. موربیدیتی فیزیکی و روانی قابل توجه نیز ممکن است خصوصاً در موارد شدید، با ناهنجاریهای اندامها مرتبط باشد.
جنسیت
سندرم هولت-اورام در دو جنس زن ومرد از نظر شیوع یکسان است.
سن
سندرم هولت-اورام به عنوان یک بیماری مادرزادی به هنگام تولد حضور دارد. درگیری دقیق اندامها ممکن است تا مدتها از نظر بالینی ظاهر نشود؛ تا زمانی که علائم قلبی بیماری تظاهر یابند یا فرد صاحب فرزندی با تظاهر شدیدتر سندرم گردد. بیماری هدایتی قلبی با افزایش سن پیشرونده است. افراد میانسال نیز اغلب با بلوک دهلیزی-بطنی شاخص یا فیبریلاسیون دهلیزی مراجعه مینمایند.
شرححال
بیماران ممکن است سابقهی فامیلی مالفورماسیون قلبی یا اندامها را ذکر کنند. همچنین ممکن است بیمار در نوزادی، مالفورماسیون واضح اندامها یا علائم نارسایی قلبی ثانویه به ASD و VSD یا بیماری هدایتی قلبی داشته باشد.
معاینهی فیزیکی
دفورمیتی اندام فوقانی شامل موارد زیر است:
- همیشه وجود دارد اما ممکن است یک طرفه یا دو طرفه باشد
- ناهنجاریهای سمت چپ اغلب شدیدتر از ناهنجاریهای دست یا بازوی راست هستند
- طول ادام فوقانی نابرابر به علت آپلازی، هیپوپلازی، فیوژن یا تکامل غیرعادی استخوانهای رادیال، کارپال و تنار
- پروناسیون و سوپیناسیون غیرعادی سادی
- عدم وجود شست یا شست تریفالانژیال
- مختل و غیرعادی بودن حرکت آپوزیشن شست
- فوکوملیا
درگیری قلبی شامل علائم زیر میباشد:
- برادیکاردی
- نبض نامنظم (اکتوپی)
- فیبریلاسیون دهلیزی
- دو جزء شدن (splitting) ثابت و پهن صدای دوم قلبی
- سوفل در گردش سیستولیک ریوی
- سوفل هولوسیستولیک (افزایش احتمال VSD)
آنومالی در موارد زیر نیز میتواند احتمال سندرم هولت-اورام را مطرح سازد:
- استخوان اولنار
- اندامهای تحتاین
- کلیهها
- چشمها
- سیستم شنوایی
- کرانیوفاشیال (جمجمه و صورت)
- مهره (ممکن است در سندرم هولت-اورام درگیر شده یا نشود)
اتیولوژی
سندرم هولت-اورام یک ناهنجاری ژنتیکی اتوزومی غالب و با نفوذ بالا است. مطالعات اولیه نشان میدهند نقص ژنتیکی واقع در بازوی بلند کروموزوم ۱۲ است. مطالعات ژنتیک مولکولی نیز بیانگر آنند که این بیماری در اثر جهشهای غیر فعال کنندهی فاکتور رونویسی TBX5 ایجاد میشوند. حالت اسپورادیک بیماری احتمالاً ناشی از جهش دنوو TBX5 در ردهی سلولهای جنسی است. شناسایی افراد مبتلا به حالت اسپورادیک بیماری که احتمالاً بیماری را به فرزند خود منتقل خواهند کرد، حائز اهمیت است.
تبیین نقش TBX5 در سندرم هولت-اورام بیانگر نقشی مهم و در عین حال نامعلوم برای TBX5 در دیوارهبندی قلبی انسان و تشکیل سپتومها، ایزومریزاسیون و تکامل اندامهای فوقانی است.
ملاحظات تشخیصی
بیماریهای ساختاری قلب یا احتمال آریتمی را در نظر بگیرید.
مشاورهی ژنتیک مناسب درخواست کرده یا بیمار را از ماهیت وراثتی ناهنجاری مطلع سازید.
پروفیلاکسی آنتیبیوتیکی مناسب در نظر داشته باشید.
کودکانی که ناهنجاری خیلی جزئی اندام داشته باشند، امکان دارد به اشتباه سالم در نظر گرفته شوند. با این حال، همهی فرزندان یا خواهر و برادران فرد مبتلا، حتی آنان که معاینهی فیزیکی اندامهایشان نرمال باشد، باید تحت اکوکاردیوگرافی و رادیوگرافی اندام فوقانی قرار بگیرند.
سایر ملاحظات
در بیماران مشکوک به سندرم هولت-اورام موارد زیر را نیز در نظر داشته باشید:
- آنومالی کروموزومی
- سندرم دوئن به همراه نقص radial ray (سندرم اوکی هیرو)
- آنمی فانکونی
- مواجهه با تراتوژنها
- ترومبوسیتوپنی بدون سندرم رادیوس (TAR)
- سندرم Townes-Brocks
- سندرم Ulnar-mammary
- نقاص مهرهای، آترزی آنال، فیستول تراکوازوفاژیال یا آترزی ازوفاژیال و کمپلکس آنومالیهای کلیوی و رادیال (کمپلکس VATER )
تشخیص افتراقی
- نقص سپتال دهلیزی
روند تشخیصی
رادیوگرافی
رادیوگرافی مچ
درگیری اندام در برخی موارد با معاینهی فیزیکی مشخص میشود. اگر درگیری در ظاهر واضح نباشد، رادیوگراف اندام فوقانی و دست تهیه کنید تا آنومالیهای جزئی استخوانهای مچ، چنانچه در تصویر زیر مشاهده میشود، آشکار گردد.
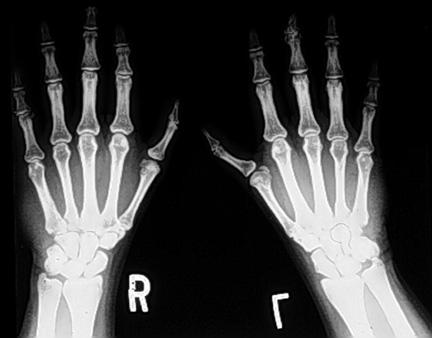
رادیوگراف خلفی-قدامی دستان یک بیمار مبتلا به سندرم هولت-اورام. فالانکس دیستال شست چپ هیپوپلاستیک است. استخوانهای کارپال هر دو دست غیرطبیعی هستند؛ اما ناهنجاریها در سمت چپ شدیدتر از سمت راست است. درواقع، ناهنجاریهای radial ray اندام فوقانی سمت چپ اغلب نسبت به سمت راست شدیدتر هستند. اسکافوئید و تراپزیوم دست چپ بزرگ شده و شکل غیرطبیعی دارند که موجب جابجایی شست به موقعیت دیستال شده است. به اختلالات مشخص شده در کاپیتیت و همیت چپ دقت کنید. استایلوس استخوان رادیوس سمت چپ نیز مسطح شده است.
افرادی که در استخوانهای رادیال پرهآگزیال ناهنجاری نداشته باشند، به سندرم هولت-اورام مبتلا نیستند.
رادیوگرافی سینه
یافتهها ممکن است نشاندهندهی بزرگ شدگی شریانهای ریوی به علت هایپرتنشن ریوی یا کاردیومگالی باشند. شواهد مبتنی بر نارسایی احتقانی قلب نیز ممکن است وجود داشته باشد.
اکوکاردیوگرافی
اکوکاردیوگرافی روش انتخابی جهت تعیین نقایص سپتال یا سایر آنومالیهای قلبی است. شایعترین آنومالی قلبی، ASD در دهانهی سکوندوم است. برخی بیماران همچنین ممکن است تنها VSD داشته باشند. بزرگشدگی قابل توجه و غیر قابل توضیح دهلیز راست در جنین ممکن است نشان دهندهی سندرم هولت-اورام باشد؛ در این شرایط، اندامهای فوقانی را از نظر ناهنجاریها بررسی کرده و با والدین، در مورد ژنتیک جنین گفتگو کنید.
افرادی که به حالت شدید بیماری دچار باشند، امکان دارد VSDهای متعدد داشته باشند (سپتوم Swiss-cheese). سایر آنومالیهای قلبی نیز شامل ایزومریسم غیرطبیعی و آنومالی برگشت ورید ریوی است. اگرچه انواع متعدد و نادری از کمپلکس بیماریهای مادرزادی قلب ممکن است با سندرم هولت-اورام مرتبط باشند.
الکتروکاردیوگرافی و هولتر مانیتورینگ
به منظور بررسی سیستم هدایتی قلب، ECG بگیرید. در صورت وجود دیسریتمی متناوب، هولتر مانیتورینگ ۲۴ ساعته میتواند مفید واقع شود. بررسی دورهای درگیری سیستم هدایتی حتی در غیاب بیماری ساختمانی قلب مهم بوده و میتواند ماهیت پیشرفت این یافته را مشخص سازد. در ارزیابیهای ECG باید فیبریلاسیون دهلیزی در نظر گرفته شود.
بررسی ژنتیک
بررسی ژنتیک حائز اهمیت است. بیمارانی را که احتمال سندرم هولت-اورام در موردشان مطرح است، برای ارزیابی به کاردیولوژیست یا متخصص ژنتیک مجرب در زمینهی بیماریهای وراثتی قلبی-عروقی معرفی کنید. سابقهی فامیلی کامل و با جزئیات از بیمار تهیه کنید تا مطمئن شوید جهش جدیدی در کار بوده یا این که جزئی از یک سندرم خانوادگی است. به منظور تعیین خانوادگی یا اسپورادیک بودن ماهیت سندرم در خانواده، رادیوگرافی مچ برای والدین بیمار مبتلا به سندرم هولت-اورام درخواست کنید.
آنالیز TBX5 از نظر جهشها در روند معمول کلینیکی انجام نشده و همچنان یک ابزار تحقیقاتی است. جهشهای TBX5 در تقریباً ۷۵% بیماران دارای کرایتریای بالینی قطعیِ سندرم هولت-اورام دیده میشود. بررسی مورد به مورد اختصاصی بیماران از نظر همبستگیهای ژنوتیپی-فنوتیپی امکانپذیر نیست. قابلیت شناسایی جهش عامل بیماری در یک خانواده، میتواند امکان گزینههای پیشرفتهی باروری را برای زوجهایی که ۵۰ درصد احتمال درگیری فرزندشان وجود دارد، فراهم کند. از این گزینهها میتوان به تشخیص ژنتیکی قبل از لانهگزینی اشاره کرد.
پروسهها
برای تعیین ماهیت و شدت شنت داخل قلبی در بیمارانی که در خطر بالا برای سندرم آیزنمنگر هستند، میتوان کاتتریزاسیون قلبی انجام داد؛ زیرا این بیماران ممکن است به مداخلهی جراحی نیاز داشته باشند.
مراقبت پزشکی
میتوان به طور معمول به صورت سرپایی ارزیابی بالینی انجام داد؛ اما گاهی بررسی در شرایط بستری و درمان با جراحی ضرورت پیدا میکند. از این رو، بیماران را از نظر قلبی-عروقی و مداخلات جراحی ارزیابی کنید. به منظور تصحیح نقایص قلبی یا احتمالاً بهبودبخشی به عملکرد اندامها نیز میتوان از درمان جراحی استفاده کرد. داروی خاصی برای این بیماری تعریف نشده است. اگرچه بسته به شدت بیماری قلبی مادرزادی، نیاز به پروفیلاکسی آنتیبیوتیکی و داروهای ضدانعقاد محتمل است. همچنین ممکن است برای ارزیابیهای تشخیصی و مداخلات جراحی، بیمار جابهجا شده و انتقال داده شود.
مشاورهها
با متخصصان زیر مشاوره کنید:
- کاردیولوژیست
- متخصص ژنتیک
- جراح قلب و قفسهی سینه
- جراح ارتوپدی
فعالیت و رژیم غذایی
رژیم غذایی خاصی نیاز نیست. در صورت وجود درگیری قلبی ناشی از بیماری یا نارسایی قلبی، فعالیتهای بیمار را محدود کنید.
پیشگیری
هیچ فاکتور محیطی شناختهشدهای به عنوان عامل بیماری وجود ندارد. از این رو، پرهیز خاصی لازم نیست.
مانیتورینگ طولانی مدت
بیماران مبتلا به بیماری قلبی مادرزادی شاخص باید حداقل به طور سالانه فالوآپ شوند. در مورد بیماری هدایتی قلبی، فالوآپ دورهای تمامی بیماران الزامی است.
مراقبت جراحی
بسیاری از نقایص قلبی همچون ASD و VSD، در صورت عدم وجود هایپرتنشن ریوی یا نارسایی بطنی، با جراحی کاملاً قابل اصلاح هستند. ابزارهای زیرجلدیِ ترانس کاتتری متعددی در حال حاضر مورد تأیید سازمان غذا و داروی آمریکا بوده و ممکن است در آینده گزینههای غیرجراحی نیز مطرح شوند.
نقایص سپتالی که با شنت قابل توجه همودینامیک همراه نباشند، نیاز به اصلاح ندارند. کودکان مبتلا به آنومالیهای شدید اندام را میتوان به منظور اجرای پروسههایی همچون پولیسیزاسیون انگشت پنجم (با هدف بهبود عملکرد اندام فوقانی) به جراحان ارتوپدی ارجاع داد. از اندام مصنوعی نیز احتمالاً بتوان برای کودکان مبتلا به کوتاهی شدید اندام استفاده کرد.
درمان دارویی
هیچ دارویی در درمان نقایص آناتومیک بیماران مبتلا به سندرم هولت-اورام، مؤثر نیست. طبق گایدلاینهای استاندارد مجمع قلب آمریکا و کالج کاردیولوژی آمریکا، برای بیماران مبتلا به بیماری مادرزادی قلبی باید پروفیلاکسی آنتیبیوتیکی صورت گیرد. برای بیماران مبتلا به هایپرتنشن ریوی هم بایستی داروی ضدانعقاد تجویز شود. در بیماران مبتلا به فیبریلاسیون دهلیزی نیز حتماً باید کاردیوورژن و دارودرمانی آنتی آریتمیک یا ضدانعقادی در نظر گرفته شود.
فالوآپ
آموزش بیمار
مطمئن شوید اعضای خانوادهی بیمار از این که ناهنجاری مذکور اتوزومی غالب بوده و ۵۰ درصد احتمال ابتلای فرزند بیمار وجود دارد، آگاهی داشته باشند. توضیح دهید که شدت یک ضایعه در والد، نشاندهندهی پتانسیل شدت آن در فرزند نیست. برای آموزش بیمار نیز میتوان همچون نقص سپتال بطنی یا فیبریلاسیون دهلیزی عمل کرد.