شما محصولی از ژنهای هر دوی والدینتان نیستید. حک گذاری ژنومی (Genomic imprinting)، فرایندی است که طی آن تنها یکی از دو نسخه ژنها بیان میشود و ممکن است در ترکیب با جهشها به بیماری منجر شود.
مقالات مرتبط:
هر دو والد به میزان یکسانی محتوای ژنتیکی خود را به فرزندان منتقل میکنند. با این حال، پدیدهای در طی رشد به نام حک گذاری ژنومی، گاها منجر به بیان گستردهتر ژنهای خاصی از یکی از والدین میگردد. این فرایند نخستین بار در سال ۱۹۸۴، و زمانی که دو آزمایشگاه نشانه یا imprinitی را یافتند که موجب تمایز ژنهای خاصی در کروموزومهای پدری و مادری شده و منجر به بیان تنها یکی از نسخههای موجود از این ژنها میگردد، توصیف شد. بیان ژنهای موجود در ناحیه حکگذاری شده واقع در ژنوم ارگانیسم، بستگی به والد منشا ژن دارد. درنتیجه، وراثت هر دو نسخه پدری و مادری ژنها، برای پیشرفت فرایند طبیعی رشد لازم است.
به منظور درک نحوه عملکرد حک گذاری، به جای تمرکز بر کل ژنوم، تاثیر این فرایند را بر نواحی کروموزمی کوچکتر و ژنهای منفرد در نظر بگیرید. برای بسیاری از ژنهای دیپلوئید، حتی در صورت وجود نقص در نسخه به ارث رسیده از یکی از والدین، الل جایگزین سالمی از والد دیگر وجود دارد. با این حال، در صورت وقوع حک گذاری، علیرغم وجود دو نسخه از ژن، همانند این است که فرد از لحاظ آن ژن هاپلوئید میباشد. به عبارت دیگر، الل جانشینی وجود ندارد و این امر ژنهای حک گذاری شده را به تاثیرات زیانبار جهش ها آسیب پذیرتر میکند. به علاوه، ممکن است ژنهای و جهشهایی که توارث مغلوب دارند، در صورت حکگذاری، بیان شده و الل غالب خاموش گردد.
بیماریهای مرتبط با حک گذاری ژنومی
همان طور که انتظار دارید، در اثر ایجاد جهشها و حذفها در ژنهای حک گذاری شده، وقوع بیماریها ممکن میباشد. در نتیجه در صورتی که حک گذاری ژنومی به وقوع بپیوندد، دیزومی تکوالدی یا به ارث بردن دو کروموزوم از یک نوع فقط از یکی از والدین میتواند منجر به بیماری شود. به علاوه، در صورت وجود جهش در ژنهای مسئول فرایند حک گذاری و عدم تنظیم صحیح فرایند حک گذاری نیز ممکن است بیماری ایجاد شود. برخی از بیماریهای ژنتیکی مرتبط با وقوع خطا در حک گذاری ژن ها و نواحی کروموزومی خاص، عبارتند از: سندروم پرادرویلی، سندروم آنجلمن و انواعی از سرطانها.
سندروم پرادرویلی و سندروم آنجلمن
سندروم پرادرویلی (Prader-Willi syndrome) نخستین بار در سال ۱۸۸۷ و توسط John Langdon Down (وی همچنین نخستین فرد توصیف کننده سندروم دارون بود) توضیح داده شد. بعدها در سال ۱۹۵۶، Andrea Prader، Alexis Labhart و Heinrich Willi این بیماری را گزارش کردند. شیوع سندروم پرادرویلی، یک مورد از هر ۲۰۰۰۰ تولد است و با اختلالات رفتاری و شناختی متعددی از جمله عقبماندگی ذهنی، نقایص رشد جنسی، هایپرفاژی و چاقی در ارتباط میباشد.
در سال ۱۹۶۵، دکتر هری آنجلمن، برای نخستین بار علائم سندروم آنجلمن را گزارش نمود. این اختلال تقریبا در یک مورد از هر ۱۵۰۰۰ تولد رخ میدهد و با اختلالات رشد، عقبماندگی ذهنی، اختلالات خواب، تشنج، آتاکسی، بیش فعالی و خلق شاد و خندههای بیش از حد، شناخته میشود.
سندرم پرادرویلی و آنجلمن، نخستین بیماریهای ناشی از حک گذاری ژنومی بودند که در انسان شناسایی شدند. علائم این دو اختلال، بسیار متفاوت از یکدیگرند؛ اما دانشمندان دریافتهاند که هر دو بیماری درنتیجه حذفهایی غیرقابل تمیز در کروموزم ۱۵ (ناحیه ۱۵q11-q13) رخ میدهند. آنچه باعث تمایز این دو اختلال از یکدیگر میشود، منشا پدری کروموزوم درگیر است. سندروم پرادرویلی، در نتیجه از دست رفتن گروهی از ژنهای واقع در کروموزوم ۱۵ رخ میدهد که از پدر به ارث رسیدهاند. در مقابل، سندروم آنجلمن در اثر از دست رفتن همین ژنها در کروموزوم ۱۵ به ارث رسیده از مادر رخ میدهد. از آن جایی که ناحیه حک گذاری شده موردنظر در کروموزوم ۱۵ حاوی ژنهایی است که هم در نسخه پدری و هم در نسخه مادری میتوانند بیان شوند، Joan Knoll و همکارانش نتیجه گرفتند که هر دو سندروم پرادرویلی و آنجلمن درنتیجه نقایصی در ژنهای حکگذاری شده رخ میدهند.
در بیشتر موارد (۶۰-۷۰ درصد)، سندروم پرادر-ویلی در نتیجه جهش در نواحیای حاصل میشود که در ژن small nuclear ribonucleoproteinpolypeptide N، ژن necdin و سایر ژنهای احتمالی رخ میدهند. در ۲۰-۳۰ درصد بقیه بیماران، اختلال به علت وجود دو نسخه از کروموزوم مادری ۱۵ و نبود نسخه پدری حاصل میشود. این حالت، دیزومی تک والدی نامیده میشود. محققان هنوز نمیدانند که چرا عدم بیان این ژنهای حک گذاری شده پدری، منجر به سندروم پرادرویلی میگردد.
خطاهای ژنتیکی مرتبط با سندروم آنجلمن متفاوت هستند. Tatsuya Kishino و همکارانش نشان دادند که سندروم آنجلمن به علت نبود بیان ژنی که به صورت مادری بیان میشود، در همان ناحیه رخ میدهد که UEB3A نام دارد. ژن UEB3A پروتئینی به نام E3 ubiquitin ligase را کد میکند که در هدفگیری پروتئینها به منظور تجزیه آنها نقش دارد و تنها در مغز حک گذاری می شود. از دست رفتن UEB3A منجر به تجزیه ناهنجار پروتئینها هنگام رشد مغز و درنتیجه سندروم آنجلمن میگردد. در واقع، در بسیاری از موارد (۶۵-۷۰ درصد)، سندروم آنجلمن در نتیجه جهشهای حذفی به ارث رسیده از مادر در ژن UEB3A رخ میدهد. با این حال، این بیماری میتواند در نتیجه دیزومی تک والدی پدری ، جهش در ژن UEB3A و خطاهای حک گذاری مانند نبود متیلاسیون در نسخه مادری نیز حاصل شود.
سرطان
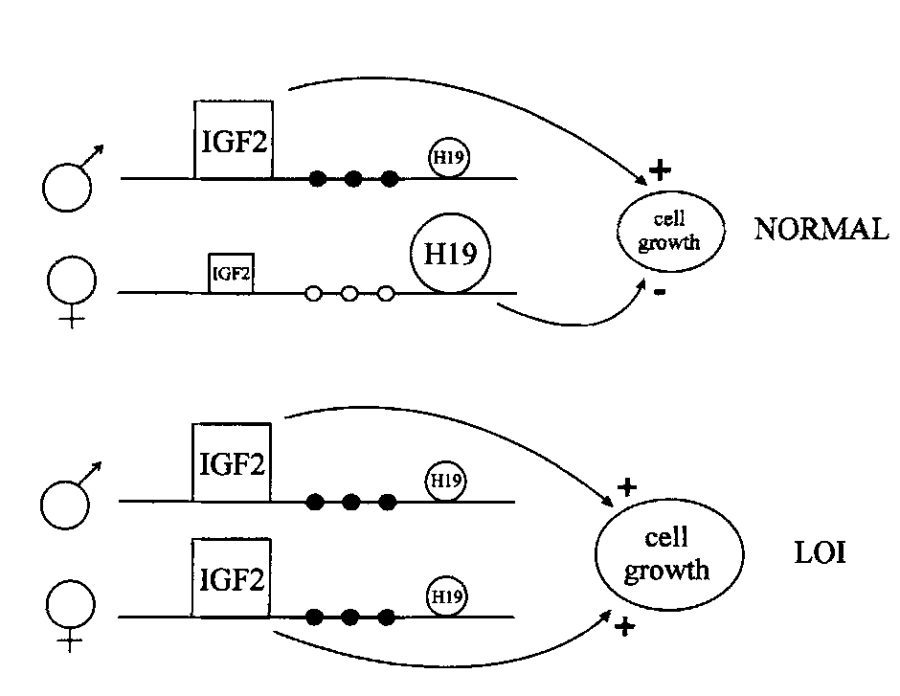
علاوه بر این دو سندروم، حک گذاری ژنومی با سرطانهای مشخصی نیز مرتبط میباشد. به عنوان مثال، تومور ویلمز نوعی سرطان کلیه جنینی است که با لوکوس IGF2/H19 در کروموزوم ۱۱ در ارتباط میباشد. H19 نوعی RNA غیرکدکننده با عملکردی نامعلوم است که میتواند از رشد جلوگیری کند. IGF2، فاکتور رشد شبه انسولین ۲ را کد میکند که بیان آن در بسیاری از تومورها بالا است. این دو ژن هر دو حک گذاری شدهاند و به طور معمول تنها نسخه مادری H19 و نسخه پدری IGF2 بیان میشود. در سلولهای سرطانی، هر دو ژن H19 و IGF2 حک گذاری خود را از دست میدهند. فرضیه محققان این ست که در صورتی که عملکرد H19 خاموش کردن بیان IGF2 باشد، در نتیجه از دست رفتن بیان H19 میتواند باعث بیان بیش از حد IGF2 شده و تومورزایی را باعث شود.
پژوهشهای انجام شده نیز این نتیجهگیری را تایید میکنند و سلولهای تومور ویلمز، حک گذاری کروموزم مادری خود را از دست داده و به الگوی پدری متیلاسیون سویچ کردهاند. به علاوه، Thomas Moulton و همکارانش نشان داده اند که بیان H19 mRNA در بسیاری از تومورهای ویلمز، حداقل به میزان ۲۰ برابر کاهش مییابد که نشان از غیرفعال شدن بیان H19 در سلولهای توموری دارد. این امر منجر به بیان بیش از حد IGF2 و کاهش H19 میگردد. از آنجایی که عملکرد H19 در آهسته کردن رشد سلولی و IGF2 رشد سلولی را تحریک میکند، از دست رفتن حک گذاری لوکوس H19/IGF2 ، منجر به رشد سلولی خارج از کنترل و تشکیل تومور خواهد شد.
نبود حک گذاری ژنومی (نبود بیان ژن نرمال اختصاصی الل)، در صورت فعال شدن اللی که معمولا خاموش است و میتواند باعث رشد سلول شود، منجر به سرطان و رشد و تقسیم خارج از کنترل سلولها میگردد. به عنوان مثال، نبود حک گذاری ژن IGF2، علاوه بر تومور ویلمز، با انواع فراوان دیگری از سرطان مانند سرطان ریه، کولون و تخمدان نیز در ارتباط است. سرطانها میتوانند در صورت حک گذاری ژنهای سرکوب کننده تومور نیز رخ دهند و نسخه ای از ژن بعدی که به صورت منفرد بیان میشود، دچار جهش شده و یا عملکرد خود را از دست میدهد.
اختلالات حک گذاری ژنومی رو به افزایش هستند
در سالهای اخیر، شاهد وقوع افزایش در بروز انواع مختلف بیماریهای مرتبط با حک گذاری ژنومی ، خصوصا در کودکان متولدشده با استفاده از تکنولوژیهای کمک باروری مانند IVF بوده ایم. از آنجایی که حک گذاری در طی گامتوژنز رخ میدهد، محققان حدس میزنند که بخشهای مختلفی از فرایند کمکباروری، مانع از حک گذاری درست ژنها و یا انتقال پایدار آن ها طی مراحل اولیه تکامل جنینی میشود. درک فرایند حک گذاری و تعیین شرایطی که با فرایند حک گذاری نرمال تداخل میکند، میتواند به کاهش بروز این بیماریها کمک کند.