نمونههایی از روشهای مبتنی بر ژن درمانی برای بهبود چاقی و اختلالات مرتبط با چاقی
اساس ژنتیکی چاقی باعث تلاشهای قابل توجهی برای توسعه ژن درمانی در معالجه و پیشگیری از چاقی است. تلاشهای بسیاری برای تهیه نسخه کپی از ژن عملکردی برای سلولها صورت گرفته تا پروتئین عملکردی از دست رفته به علت نقص ارثی جبران شود. درمقابل، رویکردهایی نیز در تلاش برای کاهش سطح یک محصول ژنی مسئول چاقی است. جدول ۲ یک خلاصه کوتاه از چند مثال که در چند سال گذشته ظهور کرده، ارائه میدهد.
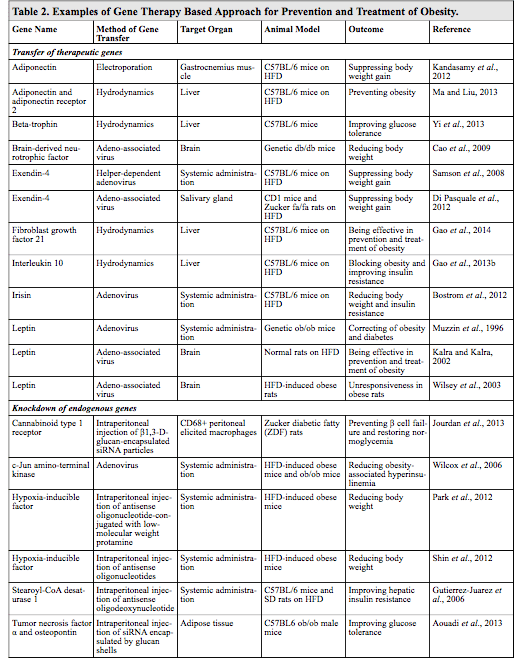
نقص ژن ob در لپتین با چاقی شدید ارتباط دارد. Muzzin و همکارانش نشان دادند انتقال ژن لپتین نوترکیب با واسطه آدنوویروس (Ad) در موشهایی با ژنتیک ob/ob باعث اصلاح چاقی و دیابت میشود. علاوه بر تجویز سیستمیک، انتقال ژن لپتین به سیستم عصبی مرکزی نیز مورد مطالعه قرار گرفت: با این حال، اثرات متمایزی در مدلهای مختلف حیوانات ایجاد شد. Kalra و همکارانش با استفاده از یکبار تزریق ویروس نوترکیب مرتبط به آدنو (AAV) حامل ژن لپتین به سومین مغز استخوان موش صحرایی بالغ که با HFD تغذیه میکند، اثبات کردند انتقال ژن مرکزی لپتین توانایی ایجاد اثرات مفید در طولانی مدت برای حفظ وزن بدن و بهبود متابولیسم سیستمیک را دارند، که احتمالا به دلیل کاهش مصرف غذا و افزایش همزمان مصرف انرژی است. Wilsey و همکارانش با فرایندی مشابه اثرات درمانی انتقال مرکزی ژن لپتین با واسطه AAV در موشهای چاق ناشی از HFD را ارزیابی کرد. بااینکه موشهای صحرایی نرمال به غذای معمولی به این درمان به شدت پاسخ دادند و کاهش وزن قابل توجهی داشتند، موشهای چاق ناشی از HFD پاسخگو نبودند، که احتمالا ناشی از کاهش گیرنده لپتین بود. درواقع، Morton و همکارانش در مطالعه افزونتری نشان دادند؛ انتقال مرکزی ژن رسپتور لپتین، وزن بدن موشهای Kolestsky را به شدت کاهش میدهد، که از مفهوم «حساسیت لپتین در مغز، در تاثیرات درمانیاش در چاقی بسیار مهم است.» حمایت میکند.
پپتید شبه گلوکاگون-۱ (GLP-1) یک پپتید کاهنده گلوکز اندوژن است که نقش مهمی در نگهداری هومئوستاز گلوکز بعد از غذا و تنظیم اشتها دارد.مطالعات جدید نشان میدهند که اثر GLP-1 در افراد چاق حتی آن دسته که میزان تحمل کلوکز عادی دارند، مختل شده است. در این زمینه، استفاده از یک وکتور کمکی مستقل آدنوویروسی به Samson و همکارانش کمک کرد تا اثر درمانی بیان طولانی مدت exendin-4 ، یک پلیپپتید آگانویست رسپتور GLP-1، در موشهای C57BL/6 تغذیه شده با HDF را ارزیابی کنند. اطلاعات آنان نشان داد انتقال ژن exendin-4 افزایش وزن بدن را در موشهای تغذیه شده با HDF سرکوب میکند، درنتیجه منجر به بهبود همئوستاز گلوکز میشود که تاحدی هم همراه با افزایش مصرف انرژی بود. افزون بر این مطالعات، در تحقیقاتی توسط Di pasquale و همکارانش، بیان expendin-4 با واسطه AAV در غدد بزاقی، افزایش وزن بدن را سرکوب کرده و مقاومت به انسولین را در موشهای CD1 و موشهای چاق Zucker fa/fa که با HDF تغذیه میشوند را کاهش میدهد.
مقالات مرتبط: ژن درمانی چاقی
فاکتور نوروتروفیک مشتق شده از مغز (BDNF) نیز یکی از اعضای خانواده «نوروتروفین» از فاکتورهای رشد است که در درجه اول در تمایز، رشد و بقا نورون اعمال میشود. نقص BDNF باعث چاقی شدید هایپرفاژیک میشود. Nakagawa و همکارانش همواره، از تزریق داخل صفاقی پروتئین نوترکیب BDNF در موشهای C57BL/6 چاق در نتیجه HDF و موشهای KKAt چاق ژنتیکی، استفاده میکردند. که نشان داد تجویز اگزوژن پروتئین BDNF قادر به کاهش افزاینده مصرف غذا است، در نتیجه باعث ایجاد اثرات ضدچاقی و ضد دیابت میشود. به همین ترتیب، انتقال ژن BDNF قادر به تولید اثرات مفید مشابه در متابولیسماند. برای مثال، Cao و همكارانش از انتقال ژن BDNF به واسطه AAV در مغز استفاده كردند. این کار منجر به توليد کاست خود تنظیم شونده فیزیولوژیکی شد که میتواند باعث تولید نمودار بیان پروتئین BNDFی که به شدت کنترل میشود، که کار تقلید از مکانیسم فیدبک فیزیولوژیکی اندوژن در بدن، بانی تنظیم مجدد نقاط بازرسی هیپوتالاموسی به حالت معکوس چاقی شدن و بهبود اختلالات متابولیکی مرتبط در موشی با ژنتیک db/db را انجام میدهد.
ارزیابیهای پیش بالینی ژن درمانی فاکتور رشد فیبروبلاست۲۱ (FGF21) مبتنی بر ژن صورت گرفته است. FGF21 پروتئینی ترشحی است که نقش مهمی در تنظیم گلوکز و متابولیسم لیپید دارد. برخلاف بسیاری از اعضای دیگر خانواده فاکتور رشد فیبروبلاست، FGF21 در تکثیر و تمایز سلول عملکرد ندارد. موش فاقد FGF21 زمانیکه با HDF تغذیه شود، تسریع رشد چاقی را نشان میدهد. درمقابل، تجویز اگزوژن پروتئین FGF21 در کاهش وزن بدن موثر بوده و نشان دهندهی کاربرد بالقوه FGF21 برای درمان چاقی است. خواص دارویی پروتئین نوترکیب FGF21 نشان میدهد در موش، نیمه عمر خون کمتر از دو ساعت است. بنابراین، تکرار تزریق برای تولید مزایای پایدر در درمان چاقی و دیابت ضروری است. در این زمینه، اثر درمانی انتقال ژن FGF21 با استفاده از روشهای هیدرودینامیک در چاقی موش C57BL/6 ناشی از HDF مورد ارزیابی قرار گرفت. نتایج ما نشان داد انتقال ژن FGF21، سطح بالای پایدار از FGF21 در خون ایجاد میکند، که منجر به اثرات مختلفی از جمله کاهش چربی، کاهش کبد چرب و بهبود تحمل گلوکز میشود که با بیان ژنهای حیاتی مرتبط با ترموژنز، آدیپوژنز و التهاب مزمن مرتبط است. آدیپونکتین یک آدیپوکین مشتق شده از چربی است که در چاقی تنظیم کاهش میابد. شواهد جدید نشان میدهند که اختلال ژنتیکی ژن آدیپونکتین باعث افرایش جاقی ناشی از HDF در نوشهای C57BL/6 میشود که با کاهش اتوفاژی میتوکندری همراه است. درمقابل، انتقال ژن بواسطه الکتروپوراسیون از DNA پلاسمید آدیپونکتین موش به عضله gastrocnemius موش C57BL/6 به طور قابل توجهی افزایش وزن بدن در اثر رژیم را سرکوب میکند. از آنجاییکه رسپتور آدیپونکتین هم در چاقی تنظیم کاهش میابد، ما همزمان ژنهای آدیپونکتین و رسپتور آن در موشهای AKR/J تغذیه شده با HFD را منتقل میکنیم و ثابت کردیم این استراتژی در بلوکه کردن چاقی و مقاومت به انسولین مرتبط با چاقی موثر است. درمان با ژن آدیپونکتین به دلیل خواص مستعد تجمع آدیپونکتین نوترکیب به پروتئین درمانی ترجیح داده میشود.
ایریسین یک پروتئین ترشحی است که به عنوان پیامرسان بین عضله و بافت چربی عمل میکند. بیان بیش از حد آیریسین از طریق انتقال ژن بواسطه Ad در موش چاق ناشی از HFD وزنبدن را به آرامی کاهش میدهد و تحمل گلوکز را به شدت بالا میبرد که در وهله اول منجر به قهوهای شدن چربی سفید و بالا رفتن مصرف انرژی میشود. اخیرا مطالعهای توسط Lee و همکارانش نشان داد همچنین قرار گرفتن در معرض سرما هم میتواند منجر به افزایش سطح اریسین در گردش شود، پس ترشح این پروتئین تکامل یافتهتر از انقباض عضله مرتبط با لرزش است. از نظر مکانیکی، اریسین باعث فسفریلاسیون پروتئین کیناز p38 با میتوژن فعال شده است و کیناز مرتبط با سیگنال خارج سلولی، مسیر را علامتدهی میکند، در نتیجه تنظیم افزایشی UCP-1، وزن بدن را کاهش میدهد و هومئوستاز گلوکز را بهبود میبخشد.
شواهد بسیاری حاکی از آن است که التهاب مزمن به عنوان یک بازیکن اصلی در چاقی و اختلالات متابولیکی مرتبط با چاقی است. التهاب مزمن همراه باچاقی در وهله اول از ماکروفاژهای نفوذی جربی گرفته شده است. مهمتر از همه، سرکوب التهاب مزمن با تخلیه ماکروفاژهای چربی از چاقی جلوگیری کرده و متابولیسم سیستمیک در موشهای C57BL/6 تغذیه شده با HFd را بهبود میبخشد. برای بلوکه کردن التهاب ناشی از رژیم و همراه با چاقی، ما اثر انتقال هیدرودینامیک مبتنی بر ژن IL-10 در موش C57BL/6 تغذیه شده با HFD را بررسی کردیم، مطابق مطالعات گذشته، نتایج ما واضحا نشان میدهند که انتقال ژن IL-10 به طور قابل توجهی باعث بهبود مقاومت انسولین و تحمل گلوکز در موشهای مذکور میشود. جالب توجه است، علاوه بر مزایای آن در متابولیسم گلوکز، شامل بیان بیش از حد IL-10 در سرکوب افزایش وزن در تغذیه شدگان با HFD نیز هست، که همراه با سرکوب نفوذ ماکروفاژهای چربی و سرکوب التهاب مزمن است.
یک عارضه متابولیکی شایع از چربی، مقاومت انسولین است، که نتیجهاش اختلال عملکرد و آپوپتوز سلولهای بتای پانکراسی است. افزایش تکثیر سلولهای بتای پانکراس نیز چالش دیگری در درمان دیابت ناشی از چربی است. مطالعهای اخیرا توسط Yiو همکارانش، ثابت کرد بتا-تروفین در تسریع روند تکثیر سلول بتای پانکراس موثر است. بتا-تروفین یک پروتئین ترشح شده است که بطور عمده توسط کبد تولید میشود و به عنوان یک پیامرسان برای ارتباط بین کبد و پانکراس استفاده میشود. اگرچه در تحقیقات Wang، بتا-تروفین در تنظیم مقادیر متابولیسم تری گلیسیرید دخیل بوده و موشهایی با کمبودیتا-تروفین اختلال در هومئوستاز گلوکز نشان ندادهاند، دادههای داخل بدن تهیه شده توسط Yi ظاهرا نشان میدهد که بیان بیش از حد بتا-تروفین توسط روش هیدرودینامیک مبتنی بر تکثیر سلولهای بتای پانکراس، منجر به افزایش انسولین گردش خون و بهبود تحمل گلوکز در موش میشود. این داده حاکی از پتانسیل کاندیداتوری ژن بتاتروفین در ژن درمانی برای دیابت وابسته به التهاب است.
رویکرد خاموش کردن ژن مبتنی بر siRNA از طریق انتقال ژن برای کاهش بیان ژن، از نظر اثربخشی آن در درمان چاقی مورد ارزیابی قرار گرفته است. برای مثال، c-Jun amino-terminal kinase (JNK) فعال شده با استرس، نقش گرداننده در چاقی و دیابت را بازی میکند. افزایش فعالیت JNK در بافتهای هدف انسولین در هر دو مدل موشهای چاق در اثر ژنتیک یا تغذیه با HFD دیده شده است. اختلال ژنتیکی JNK1 موش را در مقابل چاقی ناشی از HFD و دیابت محافظت میکند، که نشان میدهد JNK1 واسطه اصلی چاقی است. علاوه بر این، فعال سازی JNK در بدن در سلولهای بتای پانکراس منجر به تحمل گلوکز در موشهای MKK7D میشود. این مطالعات نشان میدهد سرکوب بیان JNK باعث فوایدی در رابطه با چاقی و اختلالات مرتبط با آن میشود. درواقع، در مطالعهای برای اثبات این مفهوم Wilcox و همکارانش استراتژی نابودی JNK در هردوی موشهای چاق ژنتیکی و ناشی از HFD را ارزیابی کردند و دادههای آنها نشان داد Ad-mediated JNK1 shRNA بیان JNK1 را در بدن به شدت کاهش میدهد (تقریبا ۹۵%) در نتیجه منجر به کاهش ۴۵%ی غلظت انسولین پلاسما میشود، که نشان میدهد این روش برای درمان بیماریهای قلبی مرتبط با چاقی موثر است.