PCR چیست؟
PCR یا واکنش زنجیره ای پلیمراز (Polymerase Chain Reaction) در زیستشناسی مولکولی بهمنظور ساخت میلیونها کپی از قطعه DNA موردنظر، بهکارمیرود. به عبارت دیگر، PCR مانند یک دستگاه فتوکپی که میتواند از یک برگ کاغذ، نسخههای فراوانی تهیه کند، نمونههای کوچک DNA را تکثیر کرده و مقدار آنها را چندین برابر میکند. همانطور که اشاره کردیم، این تکنیک برای نخستین بار در سال ۱۹۸۳ توسط Kary Mullis، شیمیدان آمریکایی، ابداع شد و به واسطه تاثیر عظیمش بر بیولوژی، برای وی جایزه نوبل را به ارمغان آورد.
مقالات مرتبط:
پیش از ابداع PCR، کلونینگ DNA به این منظور مورداستفاده قرار میگرفت و پیشرفتهای فراوانی را نیز در علم ژنتیک پدید آورد. اما اشکال این تکنیکها در زمانبر بودن آنها و نیز نیاز بالا به نیروی انسانی بود. به منظور جبران این نقصها و افزایش سرعت مطالعه DNA، نیاز به متدهای جایگزین کلونیگ به وجود آمد؛ تا اینکه PCR که به صورت in vitro مولکولهای DNA را کلون میکرد، اختراع شد. PCR علاوه بر اینکه یک روش کلونینگ است، امکان انجام تکنیکهای دیگری که به سنجش کمی مولکولهای DNA و RNA نیاز دارند را نیز فراهم میکند.
اساس تکثیر توسط PCR، اصول همانندسازی DNA است و تعدادی از مواد اولیه پایه را که به طور معمول در آزمایشگاههای بیولوژی مولکولی به کار برده میشود، مورد استفاده قرار میدهد. طی این فرایند، مولکولهای DNA بارها و بارها در داخل لوله آزمایش توسط DNA پلیمراز تکثیر میشوند، تا اینکه مقادیر بزرگی از آنها فراهم گردد. درنتیجه میتوان هر ناحیهای از هر مولکول DNA ای را به وسیله آن تکثیر کرد، به شرطی که توالی نواحی موجود در دو طرف آن مشخص باشد.
علیرغم سادگی مفاهیم اولیه، PCR فرایندی پیچیده و با واکنشگرهای فراوان است. غلظت DNA الگو در ابتدا پایین است، اما با پیشرفت واکنش و استفاده از مولکولهای DNA تولیدشده به عنوان الگو، به طور ناگهانی افزایش پیدا میکند. غلظت سایر واکنشدهندهها مانند dNTPها و پرایمرها در طول واکنش تغییر چندانی نمیکند، در حالیکه برخی دیگر مانند DNA پلیمراز تدریجا محدود میشوند. تغییرات چشمگیری نیز در دما و pH و در نتیجه فعل و انفعالات مولکولی وجود دارد. پس PCR فرایندی بسیار پیچیده است که توانایی فراوانی در دستکاری و آنالیز DNA دارد.
مدت زمان کوتاهی پس از اختراع PCR توسط مولیس، این تکنیک انقلابی را در بیولوژي مولکولی پدید آورد و باعث افزایشی ناگهانی در سرعت پیشرفت مطالعات ژنومی شد. امروزه با استفاده از PCR قادر به استخراج هر ژنی از هر ارگانیسمی هستیم. سرعت، حساسیت بسیار بالا و robustness تکنیک PCR، آن را به تکنیکی معمول در تحقیقات آزمایشگاهی پزشکی و زیستشناسی که به این سه مشخصه نیاز دارند، تبدیل کرده است.
حساسیت بسیار بالای این تکنیک امکان تکثیر مقادیر اندک DNA را فراهم میکند؛ بهطوریکه میتواند مقدار کم DNA موجود در قطره خون بهجامانده در صحنه جرم، و یا نسخههای اندک ژنوم ویروسی موجود در خون بیمار را تشخیص دهد. به عنوان مثال، میتواند بیماریهای عفونی مانند AIDS را پیش از ظهور علائم، شناسایی کند و یا از روی تنها یک تار مو و تکههای جداشده پوست، هویت افراد را مشخص نماید. Robustness بالای این تکنیک به این معناست که میتواند مولکولهای DNA موجود در بافتهای بسیار آسیبدیده و تجزیه شده، و یا بافتهای واقع در محیطهایی که امکان استخراج از طریق متدهای استاندارد در آنها وجود ندارد، مانند نمونههای فیکسشده در فرمالین یا نمونههای باستانشناسی، را تکثیر نماید.
در برخی از موارد، PCR جایگزین تکنیکهای کلونینگ ژن بوده و در برخی موارد دیگر، ابزاری مکمل آن است. در کلونینگ ژن، قطعهای از DNA به وکتور الحاق و به درون سلول میزبان مانند باکتری Escherichia coli وارد میشود. مولکولDNA نوترکیب جدید در درون سلول تکثیر میشود و با تقسیم باکتریها تعداد سلولهای حامل آن افزایش مییابد. در نهایت، DNA های نوترکیب استخراج و در آنالیزها و یا دستکاریهای بعدی مورد استفاده قرار میگیرند. این نوع از کلون کردن ۲ الی ۳ روز زمان میبرد. PCR میتواند نتایج مشابهی را در عرض ۱ تا ۳ ساعت و داخل لوله آزمایش برای ما فراهم کند که در تستهای سرطان، بیماریهای ژنتیکی و تستهای پیش از تولد بسیار مهم است.
در مطالعه ژنهای جدید و بیماریهای ژنتیکی، غالبا نیاز به ایجاد کتابخانههای ژنی داریم که با استفاده از PCR و کلون کردن محصولات داخل یک وکتور مناسب انجام میشود. همچنین آزمایشاتی که در آنها تولید پروتئین و سپس مطالعه ساختار و عملکرد آن صورت میگیرد نیز، نیازمند بیان پروتئین در سلول میزبان هستند که آن نیز با کلون کردن ژن توسط PCR و الحاق آن به داخل وکتورهای مناسب انجام میگیرد. پس در بسیاری از موارد، PCR و کلونینگ ژن تکنیکهایی مکمل هستند و انتخاب مناسبترین استراتژی از اهمیت بالایی برخوردار میباشد.
PCR تکنیکی اساسی در فرایند توالییابی ژنوم است و در تعیین توالی DNA و مطالعات بعدی روی ژنها و محصولاتشان مورد استفاده قرار میگیرد. با استخراج ژن هدف، با استفاده از PCR میتوانیم توالی آن را به منظور کلونینگ و موتاژنز برش دهیم و یا با طراحی تستهای تشخیصی فرم جهشیافته ژنها را تعیین نماییم. تکنیک PCR امروزه در تمامی حوزههای علوم بیولوژیک به کار میرود. سادگی نسبی و امکان راهاندازی و استفاده آسان از آن، PCR را به یک تکنیک روتین آزمایشگاهی تبدیل و دسترسی آزمایشگاههای غیرمولکولی به تکنیکهای مولکولی را فراهم کرده است. امروزه متدها و کاربردهای فراوانی از PCR توصیف شدهاند و کیتها و محصولات تجاری فراوانی در زمینههای تحقیقاتی و تشخیص آزمایشگاهی در دسترس هستند که به برخی از آنها اشاره خواهیم کرد.
PCR چگونه انجام میشود؟
تکنیک PCR با بهرهگیری از اصول اولیه فرایند طبیعی سنتز DNA و همانندسازی، مولکولهای DNA را در داخل لوله آزمایش کپی میکند. در داخل سلولهای زنده، سیستمی پیچیده که متشکل از تعداد فراوانی پروتئین است، همانندسازی ژنوم را انجام میدهد: دو رشته DNA از هم باز میشوند و هر رشته مولکول مادر به عنوان الگویی برای تولید رشتههای مکمل دختر مورد استفاده قرار میگیرد. اساس این نسخه برداری، همان قانون معروف واتسون و کریک است: A همواره با T و G همواره با C جفت میشود.
همان طور که گفتیم، PCR مشابه یک دستگاه فتوکپی است و همانند این دستگاه که نیاز به موادی مثل کاغذ، جوهر و سختافزار دارد، PCR نیز نیازمند مواد اولیه خاصی است:
- نمونه DNAای که قرار است از آن کپی تهیه شود، توالی الگو نام دارد و غالبا شناخته شده است. DNA آغازگر معمولا DNA توتال دورشتهای ژنومی است که از بافتی مشخص و یا از سلولهای کشت شده تهیه میشود و میتواند به پیچیدگی ژنوم انسان و یا به سادگی پلاسمید باکتریایی باشد. توالیهای هدف نیز معمولا بخش بسیار کوچکی از این DNA را تشکیل میدهند. مقادیر اندک DNA آغازگر نیز، به علت حساسیت بسیار بالای PCR، برای انجام واکنش کافی است. همچنین میتوان از cDNA توتال که از استخراج RNA و قرار دادن آن تحت تاثیر آنزیم رونوشتبردار معکوس حاصل شده است، به عنوان DNA هدف استفاده نمود. این تکنیک RT-PCR نام دارد و در ادامه به آن خواهیم پرداخت.
- سنتز DNA توسط DNA پلیمراز نیازمند توالی کوتاهی از DNA به نام پرایمر است که مکمل توالی الگو بوده و آغازگر واکنش PCR میباشد. پرایمرها طولی در حدود ۸ تا ۲۰ نوکلئوتید دارند و به تعداد یک جفت تهیه میشوند. طراحی پرایمرها بهگونهای انجام میگیرد که به انتهای ۳ پریم رشتههای هدف متصل شوند و افزودن نوکلئوتیدها توسط پلیمراز نیز طبق قوانین همانندسازی به انتهای ۳’-OH پرایمر صورت خواهد گرفت. توالی پرایمرها تعیینکننده موقعیت دقیق توالی هدف موجود در نمونه DNA است؛ درنتیجه میتوان واکنش را حتی در صورت پیچیدگی نمونه اولیه، روی توالی الگو متمرکز نمود.
- سومین جزء، نوکلئوتیدها هستند که به صورت dNTP میباشند. به منظور موفقیت PCR، غلظت هر چهار نوع dNTP باید با یکدیگر برابر باشد. در صورت بالاتر از حد بودن غلظت dNTPها، خطای آنزیم پلیمراز افزایش یافته و در صورت پایین بودن آن، کارایی PCR کاهش خواهد یافت.
- آخرین واکنشدهنده اصلی، آنزیم Taq DNA Polymerase است که از باکتری Thermus aquaticus به دست آمده است و وظیفه کپیبرداری را بر عهده دارد. این باکتری در چشمههای آب داغ زندگی میکند و آنزیم DNA پلیمراز آن نیز همانند سایر آنزیمهایش به دناتوره شدن در برابر گرما مقاوم میباشد. همان طور که خواهیم دید، این ویژگی آنزیم در متدولوژي PCR بسیار ضروری است.
- البته وجود بافر نیز به منظور فراهم کردن شرایط مناسب واکنش الزامی میباشد. بافر معمولا به همراه آنزیم ارائه میشود. ترکیبات بافر مناسب برای Taq پلیمراز حاوی این ترکیبات است: بافر Tris-HCl، KCl، MgCl۲، ژلاتین و حلالهای غیریونی است. Tris-HCl یک بافر دوقطبی یونی است و pH آن بین ۶.۸ و ۸.۳ متغیر است. در دماهای بالا، pH پایین میآید و که برای Taq پلیمراز بهینه است. اهمیت KCl در اتصالات پرایمر و الگو است. منیزیم نیز از اصلیترین اجزاء PCR بوده و غلظت آن میتواند اختصاصیت و کارایی واکنش را تحت تاثیر قرار دهد.
مکانیسم اصلی PCR شامل سه مرحله است: دناتوراسیون حرارتی توالی الگو (مرحله دناتوراسیون)، اتصال پرایمرها (مرحله اتصال) و تولید نسخه مکمل با استفاده از DNA پلیمراز (مرحله گسترش). این مراحل بارها و بارها تکرار میشوند، تا اینکه از یک توالی الگو میلیونها نسخه یکسان تهیه شود. درنتیجه مقادیری از DNA که کوچکتر از آن بودند که قابل مشاهده باشند، تکثیر میشوند؛ به نحوی که میتوانند به وکتورها الحاق شده و یا روی ژل آگارز مشاهده گردند.

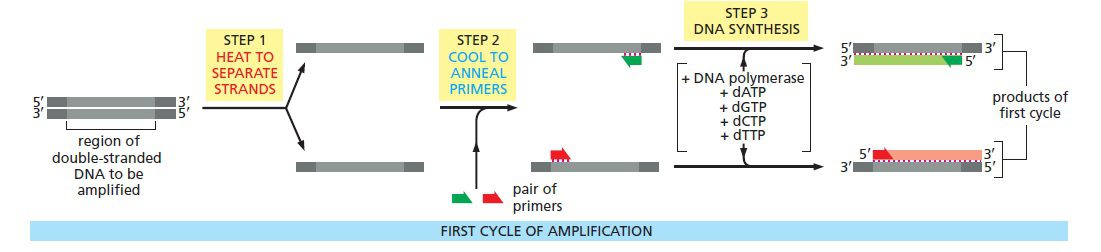
انجام متوالی این فرایندها نیازمند تغییر در دما به صورت چرخهای است؛ به عبارت دیگر مخلوط واکنش باید به طور متوالی گرم و سرد شود. این فرایند توسط دستگاه thermocycler انجام میگیرد و این فرایند نیز گاها Thermocycling خوانده میشود. thermocycler به گونهای طراحی شده است که بتواند دما را در هر کدام از بلوکهای دمایی تعیین شده، سریعا تغییر داده و در مدت زمان موردنظر در آن دما نگه دارد. در نتیجه هر چرخه میتواند در عرض چند دقیقه تکمیل گردد. این اتوماسیون یکی از مهمترین پیشرفتهایی است که به توسعه و میزان دسترسی PCR کمک کرد.
چرخه دمایی به این ترتیب است: در حدود ۹۴ درجه سانتیگراد توالی الگو دناتوره میشود (دو رشته آن از یکدیگر جدا میشوند)، در دمای ۵۰ تا ۶۰ درجه پرایمرها متصل میگردند (بسته به طول و توالی پرایمر)، و در ۷۲ درجه، Taq پلیمراز، مولکول DNA جدید را میسازد. پیش از توسعه دستگاههای thermal cycler، PCR با انتقال مخلوط واکنش به صورت دستی بین ۳ حمام آب با دماهای ۹۵، ۵۵ و ۷۲ درجه سانتیگراد صورت میگرفت که بسیار کم دقت بود.
در هر مرحله از PCR چه فرایندهایی رخ میدهد؟
مرحله دناتوراسیون
در فرایند طبیعی همانندسازی در داخل سلول، کمپلکسی چندجزئی که حاوی انواع مختلفی از آنزیمها و پروتئینها است به منظور جداسازی رشتههای DNA به کار میرود. اما در PCR این مرحله با مرحله حرارت دادن جایگزین میگردد. طی این مرحله تمام پنج جزء اصلی نامبردهشده از جمله DNA الگو حرارت داده میشوند که در نمونههای انسانی معمولا تا دمای ۹۳-۹۵ درجه سانتیگراد است. این دما به حدی بالا است که باعث از هم گسستن پیوندهای هیدروژنی بین بازها، که عامل کنارهم نگه داشته شدن رشتههای مکمل بود، شده و دو رشته DNA جداگانه را به وجود آورد. این دو رشته بهعنوان الگویی برای ساخت رشتههای جدید DNA عمل میکنند. مدت زمان افزایش دما تا این حد باید به میزانی باشد که تمام مولکولهای DNA دو رشتهای شوند. این فرایند معمولا ۳۰-۱۵ ثانیه طول میکشد.
مرحله اتصال
در این مرحله پرایمرها به رشته هدف خود متصل میشوند و زمان مورد نیاز برای آن، ۳۰-۱۰ ثانیه میباشد. به این منظور ابتدا، دما تا حدود ۶۰-۵۰ درجه سانتیگراد پایین میآید. نتیجه این کار اتصال مجدد برخی از مولکولهای تکرشتهای هدف به همدیگر و نیز اتصال پرایمرها با پیوند هیدروژنی به نقاط مخصوص به خود در DNA هدف است.
دمای اتصال باید به صورت دقیق تعیین شود، زیرا در اختصاصیت واکنش نقشی اساسی دارد. به طور معمول، هر چه دمای اتصال واکنش بالاتر باشد، واکنش اختصاصیتر خواهد بود. این دما، به نقطه ذوب پرایمر مورد استفاده بستگی دارد. دمای مورد استفاده معمولا ۵۵ درجه سانتیگراد است، اما در موارد بسیاری بهتر است که دماهای بالاتری به کار گرفته شوند. این دما در برخی واکنشها ممکن است به ۷۲ درجه سانتیگراد نیز برسد و منجر به چرخه PCR دو دمایی شود.
همانطور که گفته شد، پرایمرها توالیهای تکرشتهای DNA (یاRNA ) هستند که طولی به اندازه ۸-۲۰ باز دارند و انتهای ۳ پریم مورد نیاز برای آغاز فعالیت پلیمراز و سنتز DNA را تهیه میکنند. این مولکولها هستند که نقطه شروع فعالیت پلیمراز را در هر دو رشته از ژنوم مکانیابی میکنند؛ چون آنزیم DNA پلیمراز بازهای جدید را تنها بهDNA دو رشتهای میافزاید. پس از اتصال پرایمر، پلیمراز نیز متصل شده و ساخت رشته مکمل را آغاز میکند.
دو مولکول تکرشتهایDNA ایجاد شده در مرحله دناتوراسیون، مکمل هم بوده ولی در دو جهت متفاوت قرار دارند (منظور از جهت نحوه قرارگیری انتهاهای ۳ پریم و ۵ پریم است). در نتیجه ۲ نوع پرایمر خواهیم داشت: Forward Primer و Reverse primer. نقطه اتصال پرایمرها به گونهای طراحی شده است هر کدام به نقطه مخالف دیگری متصل شوند؛ یکی به ابتدای ژن و دیگری به انتهای آن.
مرحله گسترش
طی این مرحله، DNA پلیمراز شروع به افزودن دئوکسینوکلئوتیدها به انتهای ۳’ هر دو پرایمر کرده و مولکولهای دورشتهای جدید DNA را ایجاد میکند. به این منظور، دما به ۷۰-۷۵ درجه سانتیگراد (معمولا ۷۲ درجه) میرسد که دمای بهینه فعالیت Taq DNA پلیمراز است. درنتیجه آنزیم فعال شده و افزودن بازهای آلی و سنتز رشته جدید را از سمت ۵ پریم به ۳ پریم شروع میکند.
Taq DNAپلیمراز آنزیمی است که از نوعی باکتری گرمادوست به نام Thermus aquaticus گرفته شده است. محیط زندگی این باکتری غالبا چشمههای آب گرم است و میتواند حرارت بالای ۸۰ درجه سانتیگراد را تحمل کند؛ درنتیجه DNA پلیمراز این باکتری نیز در دماهای بالا پایدار است. این به آن معنی است که میتواند دمای بالایی را که برای جدا کردن دو رشته DNA از همدیگر در مرحله دناتوراسیون مورد نیاز است، تحمل کند. از این رو افزودن آنزیم پس از هر بار انجام شدن چرخه، مورد نیاز نخواهد بود. DNA پلیمراز بسیاری از ارگانیسمها قادر به تحمل این دما نیست. به عنوان مثال، دمای مناسب برای فعالیت پلیمراز انسانی ۳۷ درجه سانتیگراد است.
از پیشرفتهای مهم تکنولوژیک PCR، جایگزینی DNA polymerase I Klenow با DNA پلیمرازهای مقاوم به حرارت از جمله Taq پلیمراز بود. انجام مرحله گسترش در دماهای بالا، اختصاصیت واکنش را افزایش میدهد و این پلیمرازها، برخلاف Klenow در دماهای بالا غیر فعال نمیشوند. در دمای ۳۷ درجه که دمای مناسب فعالیت Klenow است، پرایمرها به قطعات غیرهدف که دارای تشابه توالی ضعیفی با توالی هدف هستند، متصل شده و اختصاصیت PCR در نتیجه تکثیر تعداد زیادی از قطعات غیرهدف، پایین میآید. استفاده از پلیمرازهای مقاوم به حرارت همچنین هزینهها را نیز کاهش میدهد؛ چرا که در هر چرخه مجبور به افزودن مجدد آنزیم نیستیم.
البته، Taq پلیمراز فاقد فعالیت اگزونوکلئازی ۳’→۵’ و درنتیجه خاصیت proofreading است. همه پلیمرازها ممکن است در اثر الحاق باز نادرست مرتکب خطا شوند و چنین اشکالاتی در همانندسازی میتواند با این خاصیت تصحیح شود. درنتیجه دانشمندان در صدد استفاده از باکتریهای مقاوم به گرمای دیگری که دارای فعالیت اگزونوکلئازی ۳’→۵’ بودند، برآمدند. از جمله این پلیمرازها، Pfu polymerase است که از باکتری Pyrococcus furiosus به دست میآید.
در فرایند گسترش، DNA پلیمراز هر دو پرایمر را تکثیر کرده و هر تک رشته الگو را به مولکولهای DNA دورشتهای تبدیل مینماید. آرایش قرارگیری پرایمرها به گونهای تنظیم میشود که جهت سنتز رشتههای جدید DNA به سمت به سمت محل اتصال پرایمر دوم باشد. رشتههای جدید میتوانند در سنتز مولکولهای بعدی به عنوان الگو عمل نمایند. درنتیجه واکنش زنجیرهای شده و افزایش نمایی در محصولات صورت میگیرد. مدت زمان مرحله گسترش بستگی به طول توالی موردنظر دارد. کپی شدن هزار باز DNA حدود یک دقیقه زمان میبرد.
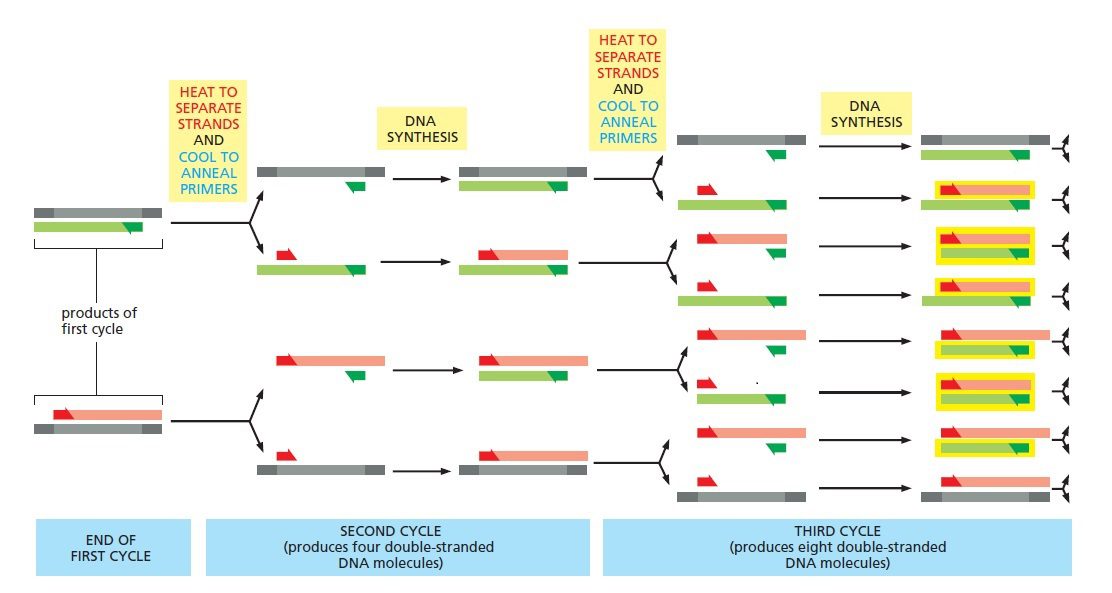
پس از اتمام نخستین چرخه همانندسازی،کل فرایند دناتوراسیون-اتصال-سنتز مجددا تکرار می گردد. در مراحل اولیه تعدادی مولکول بلند از هر کدام از مولکولهای هدف سنتز میشوند که انتهای ۵ پریم مشترک و انتهای ۳ پریم تصادفی دارند که نشاندهنده نقطه خاتمه سنتز است. در آغاز چرخه دوم، دو مولکول دورشتهای بلند حاصل در دمای بالا دناتوره میشوند؛ سپس دما افت میکند تا پرایمرها بتوانند به توالی الگو اتصال یابند و در مرحله بعدی آنزیم پلیمراز چهار رشته موجود را تکثیر میکند. در پایان این چرخه، ۴ کپی دورشتهای از توالی هدف خواهیم داشت که دو عدد از آنها بلند و مشابه محصولات چرخه اول و دوتای دیگر، مولکولهای DNA ای کاملا جدید هستند.
طی چرخه سوم است که برای نخستین بار مولکولهای با طول موردنظر به وجود میآیند. در این چرخه، مولکولهای DNA جدید تولیدشده در چرخه قبلی، منشا تولید مولکولهای کوتاهتری میشوند که انتهای ۳ پریم و۵ پریم آنها منطبق بر نقاط اتصال تعیین شده توسط پرایمرها است. پس در مراحل اولیه فرایند، تعدادی رشته بلند نیز تولید خواهند شد، اما با تکرار چرخهها در نهایت، تنها قطعه احاطه شده توسط پرایمرها تکثیر میشود و توالی الگو و محصولات اولیه PCR در اقلیت قرار میگیرند.
در طی چرخههای بعدی، تعداد مولکولهای کوتاه موردنظر به صورت نمایی افزایش یافته و در هر چرخه دو برابر میشود. درنتیجه در کارایی ۱۰۰ درصد، هر مولکول الگوی حاضر در آغاز واکنش منجر به تولید ۱۰۶ مولکول جدید پس از طی تنها ۲۰ چرخه خواهد شد. البته کارایی فرایند هیچ وقت ۱۰۰ درصد نیست و برای رسیدن به این مقدار باید تعداد چرخههای بیشتری به انجام برسند. این تعداد غالبا بین ۲۵ تا ۴۰ است و بستگی به غلظت اولیه توالی الگو، میزان خلوص آن، شرایط اولیه واکنش و کاربرد موردنظر دارد.
هر کدام از چرخهها معمولا ۵ دقیقه به طول میانجامد و تعداد دفعات تکرار در واکنش استاندارد حدود ۳۰ الی ۴۰ بار میباشد. یک آزمایش PCR میتواند در کمتر از یک ساعت به طور کامل به انجام برسد و پس از اتمام ۳۰ چرخه، حدود ۱۳۰ میلیون محصول تولید خواهد شد. این میزان معادل چندین میکروگرم محصول از چند نانوگرم مولکول DNA هدف است. البته مدت زمان انجام آن به سرعت دستگاهها نیز بستگی دارد.
در پایان، نمونه حاوی مخلوط واکنش توسط الکتروفورز در ژل آگارز آنالیز میگردد. در صورتیکه محصول موردنظر به مقدار کافی تولید شده باشد، به صورت یک نوار مشخص در الکتروفورز دیده خواهد شد. این آنالیز اطلاعات سودمندی در مورد ناحیهای از DNA که تکثیر شده است، در اختیار ما قرار میدهد. همچنین می توان محصول PCR را با تکنیکهایی مانند توالییابی تحت بررسی قرار داد.
اختصاصیت و کارایی PCR به این معنا است که تعداد بسیار اندکی از مولکولهای الگو که در آغاز PCR وجود دارند، در پایان واکنش تعداد فراوانی از مولکولهای DNA را که جرمی در حدود یک میکروگرم یا بیشتر دارند، به وجود میآورند. البته این توانایی بالای تکثیری به این معنا نیز هست که در صورت آلودگی واکنش با مولکولهای DNA دیگر از جمله محصولات واکنشهای پیشین، نتایج کاذب حاصل خواهند شد. به همین علت است که انجام واکنشهای کنترل بسیار مهم است.
پرایمرها
PCR نیازمند سنتز یک سری توالیهای الیگونوکلئوتیدی است که در سنتز DNA جدید به صورت انتخابی، به عنوان پرایمر عمل میکنند. پرایمرها در موفقیت PCR و یا شکست آن نقشی کلیدی دارند. در صورت طراحی صحیح پرایمرها، آزمایش منجر به تکثیر یک قطعه DNA واحد میگردد که منطبق بر ناحیه هدف در مولکول الگو است. در صورت طراحی نادرست، واکنش با شکست روبهرو خواهد شد. علت این موضوع، احتمالا رخ ندادن تکثیر و یا تکثیر بیش از یک قطعه است. در صورت عدم اتصال پرایمرها به نقاط مناسب، طولانی بودن فاصله بین دو پرایمر و تشکیل شدن hairpin در ساختار پرایمر به جای اتصال به توالی هدف، آنزیم Taq پلیمراز قادر به تکثیر قطعه هدف نخواهد بود. همچنین اگر هر دو پرایمر به رشته یکسانی متصل گردند، واکنش ادامه نخواهد یافت.
تعیین توالی مناسب برای پرایمر کار دشواری نیست: پرایمرها اختصاصی دو توالی بلافاصله مجاور ناحیه هدف بوده و طولی در حدود ۱۸ تا ۲۵ نوکلئوتید دارند. برای اینکه هیبریداسیون بتواند اتفاق بیفتد، هر پرایمر باید از لحاظ توالی مکمل رشته هدف باشد. همچنین انتهای ۳’ دو پرایمر مورد استفاده باید به سمت یکدیگر قرار بگیرند. سنتز پرایمرها از روی توالیهای هدف معینی صورت میگیرد، پس به این منظور باید اطلاعاتی در مورد این توالیها داشته باشیم. در حالت ایدهآل، قطعات DNA موردنظر نیز نباید طولی بیش از ۳ کیلوباز و کمتر از یک کیلوباز داشته باشند. البته قطعاتی با طول بیش از ۱۰ کیلوباز نیز میتوانند با تکنیکهای PCR استاندارد تکثیر شوند، اما این نکته را نیز باید مدنظر داشت که با افزایش طول قطعات، بازده فرایند تکثیر کاهش خواهد یافت.
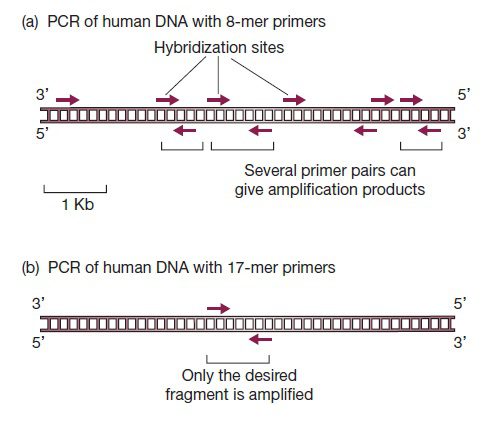
طول پرایمرها از جمله مهمترین پارامترها در سنتز پرایمر است. در صورتیکه پرایمرها کوتاهتر از حد باشند، ممکن است به نقاط غیراختصاصی متصل شده و محصولات غیراختصاصی تولید کنند. به عنوان مثال، در صورتیکه در یک آزمایش PCR، برای DNA توتال انسانی از پرایمری ۸ نوکلئوتیدی استفاده شود، به احتمال زیاد، قطعات مختلفی تکثیر خواهند شد. علت این موضوع، تعداد تکرار جایگاههای موردانتظار برای اتصال این پرایمر است که در حدود یک در ۴۸= ۶۵۵۳۶ جفتباز میباشد و در نتیجه در کل ۳۲۰۰۰۰۰ کیلوباز نوکلئوتید تشکیل دهنده ژنوم انسان، ۴۹۰۰۰ جایگاه احتمالی برای اتصال پرایمر وجود خواهند داشت. پس وجود تنها یک محصول اختصاصی در صورت تکثیر کل ژنوم انسان با پرایمری تکنوکلئوتیدی، بسیار غیرمحتمل است.
در صورت استفاده از پرایمر ۱۷ نوکلئوتیدی، فرکانس تکرار این توالی یک مورد در هر ۴۱۷= ۱۷۱۷۹۸۶۹ جفت باز است که ۵ بار بزرگتر از طول کل توالی ژنوم انسانی میباشد. پس پرایمر ۱۷ نوکلئوتیدی به احتمال زیاد تنها یک جایگاه هیبریداسیون در DNA توتال انسان خواهد داشت و یک جفت پرایمر ۱۷ نوکلئوتیدی نیز طبق انتظار، محصولی واحد و اختصاصی تولید خواهد کرد.
حال سوال این است که چرا پرایمرهای طولانیتری ساخته نمی شوند؟ طول پرایمر سرعت هیبریداسیون آن با DNA هدف را تحت تاثیر قرار میدهد و پرایمرهای کوتاهتر با سرعت کمتری هیبرید میگردند. درنتیجه کارایی PCR که بر اساس تعداد قطعات تکثیرشده در حین آزمایش تعیین میگردد، کاهش خواهد یافت. علت این موضوع ، افزایش طول پرایمرها و عدم هیبریداسیون کامل آنها با توالی الگو در مدت زمان تعیین شده در چرخه میباشد. پرایمرهای با طول بیش از ۳۰ نوکلئوتید ندرتا مورد استفاده قرار میگیرند.
در بسیاری از واکنشها هدف تکثیر یک توالی DNA واحد است؛ در این موارد، از اهداف مهم، کاهش احتمال اتصال پرایمر به نقاطی غیر از نقطه موردنظر است. پس نباید از توالیهای تکراری استفاده نمود. به منظور حصول اطمینان از مکمل نبودن بازهای داخل یک پرایمر با یکدیگر، توالی پرایمر نباید محتوی تکرارهای معکوس (inverted repeats) و یا هر گونه توالی self-complementary با طولی بیش از ۳ جفتباز باشد. به منظور جلوگیری از اتصال دو پرایمر به یکدیگر و تشکیل دایمر پرایمر، توالی ۳’ هیچ یک از پرایمرها نباید در هیچ کدام از جایگاهها مکمل خود و یا پرایمر دوم باشد.
دایمرهای پرایمر، محصول گسترش پرایمر از روی خود و یا پرایمری دیگر هستند. از آنجایی که این محصولات حاوی توالی یکی از پرایمرها و یا هر دوی آنها و نیز توالی مکمل این پرایمرها هستند، الگوی مناسبی برای واکنشهای تکثیر بعدی فراهم میکنند. نسخهبرداری آسانتر مولکولهای کوچکتر باعث بدتر شدن شرایط نیز میشود. دایمرهای پرایمر میتوانند در واکنش PCR غالب شده و پرایمر ار از توالی هدف حقیقی خود جدا نمایند.
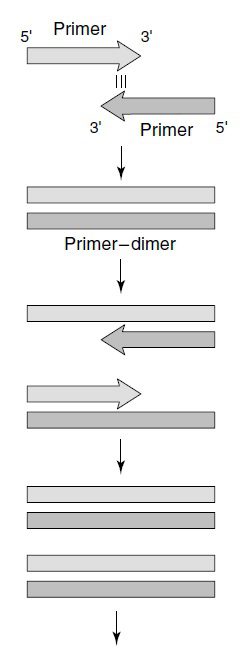
در بسیاری از موارد، احتیاجی به تطابق کامل توالی پرایمر با توالی الگو نداریم. ناحیهای که باید کاملا مکمل باشد، انتهای ۳’ است؛ زیرا این انتها است که توسط DNA پلیمراز شناسایی شده و گسترش مییابد و باید از اختصاصیت و اتصال درست آن مطمئن شد. در انتهای ۳’ حداقل سه نوکلئوتید نخست باید به طور کامل مکمل توالی هدف باشند و طی حدود ۲۰ جفتباز بعدی نیز تعداد کمی از اتصالات نادرست رخ بدهند. انتهای ۵’ اهمیت کمتری در تعیین اختصاصیت اتصال به توالی هدف داشته و امکان دستکاری در توالی به منظور تسهیل مراحل بعدی مانند کلونینگ، موتاژنز، نوترکیبی و بیان محصول، فراهم میباشد. از مهمترین تغییرات، افزودن جایگاه تشخیص آنزیم محدودکننده است. درنتیجه این کار محصول تکثیر شده میتواند به سادگی به داخل وکتور موردنظر الحاق شود.
دمای ذوب پرایمرها (Tm)
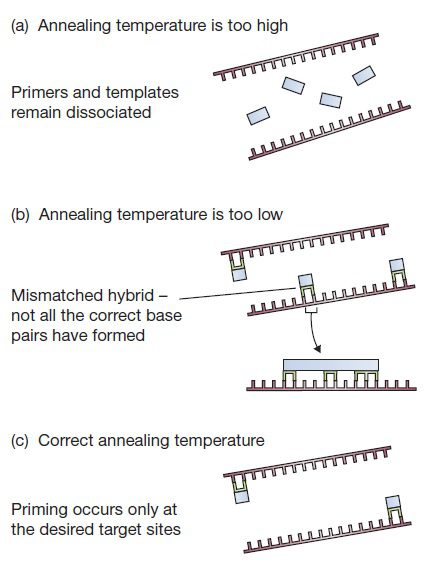
برای اینکه اتصال پرایمر به توالی مکمل موردنظر با درجه اختصاصیت بالا صورت بگیرد، دمای مرحله اتصال باید در بیشترین مقدار ممکن باشد، تا حدی که از تشکیل جفتباز بین پرایمر و محل اتصال آن جلوگیری نشود. هیبریداسیون DNA-DNA پدیدهای وابسته به دما است: در صورت بالاتر بودن دما از حد معمول، هیبریداسیون رخ نداده و پرایمرها و توالیهای الگو جدا از یکدیگر باقی خواهند ماند. در صورت پایین بودن دما نیز هیبریدهای mismatch شده پایدار خواهند بود و در نتیجه احتمال تکثیر نواحی غیر هدف بالا خواهد رفت.
معیاری سودمند برای بررسی پایداری هر دوپلکس نوکلئیکاسیدی، دمای ذوب یا Tm آن است. Tm دمایی است که منطبق بر نقطه حدواسط تغییر مولکولهای دورشتهای DNA به تکرشتهای قرار دارد و در آن جدا شدن بازهای جفتشده هیبرید رخ میدهد. بیشترین اختصاصیت اتصال پرایمر معمولا در دمایی حدود ۵ درجه سانتیگراد پایینتر از Tm محاسبه شده رخ میدهد. این دما به حدی پایین است که امکان تشکیل هیبریدی با بازهای درست جفتشده را فراهم آورد و در عین حال به حدی بالاست که از پایداری هیبریدهایی حتی با یک mismatch جلوگیری به عمل آورد. در صورتی دمای مرحله اتصال خیلی پایین تر از دمای ذوب باشد، ممکن است پرایمرها به نقاطی از DNA متصل شوند که تنها در بخشی از توالی با آن مکمل میباشند. تفاوت Tm محاسبه شده برای دو پرایمر مورد استفاده در یک واکنش نباید بیشتر از ۵ درجه سانتیگراد باشد و نیز Tm توالی هدف تکثیر شده نیز نباید بیشتر از ۱۰ درجه با پرایمرها تفاوت داشته باشد. (Gene Cloning and DNA analysis: پس دمای اتصال آزمایشهای PCR با محاسبه دمایی ۲-۱ درجه سانتیگراد پایینتر از دمای Tm پرایمرها تعیین میگردد. البته این موضوع در حالتی است که هر دو پرایمر دارای Tm یکسانی هستند. در غیر این صورت، ممکن است دمای اتصال مناسب برای یکی از پرایمرها، برای دیگری بسیار بالاتر و یا پایینتر از مقدار موردنیاز باشد.)
دمای ذوب پرایمرها و درنتیجه دمای مرحله اتصال بستگی به ترکیب بازها دارد. علت این موضوع وجود سه پیوند هیدروژنی میان جفت بازهای GC و وجود دو پیوند در بین جفت بازهای AT است. در نتیجه جدا کردن رشتههای دارای درصد بالایی از جفتبازهای GC دشوارتر از آنهایی است که محتوای GC کمتری دارند. در سنتز پرایمرها، بهینهترین حالت محتوای GC بین ۴۰ تا ۶۰ درصد و توزیع یکسان هر چهار نوکلئوتید است. این موضوع اساس فرمول زیر را که به منظور محاسبه Tm به کار میرود، تشکیل میدهد. در این رابطه [G + C] و [A + T] به ترتیب تعداد نوکلئوتیدهای G و C و A و T موجود در پرایمر میباشند.
Tm = (۴ × [G + C]) + (2 × [A + T])°C
تهیه پرایمرها
پرایمرهای الیگونوکلئوتیدی به طور گستردهای در دسترس هستند و کمپانیهای فراوانی مانند Alpha DNA، Biosource، Integrated DNA Technologies، Bio-Synthesis، Invitrogen، Midland Certified Reagent Company، MWG Biotech، PE Biosystems و Sigma Genosys، سنتز پرایمر موردنظر پژوهشگران را با هزینه پایین و در عرض چند روز پس از سفارش انجام میدهند.
در بسیاری از آزمایشهای PCR نیازمند دو نوع پرایمر با توالی متفاوت هستیم که به رشتههای مکمل خود روی توالی هدف متصل میشوند. در صورتی که توالی مولکول هدف را بدانیم، طراحی پرایمرهای مناسب به منظور تکثیر قطعه موردنیاز بسیار آسان خواهد بود. برنامههای کامپیوتری تجاری و وبسایتهای معینی به این منظور مورد استفاده قرار میگیرند. Web primer (http://seq.yeastgenome.org/cgi-bin/web-primer)، Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi)، Oligoperfect designer (http://www.invitrogen.com/content.cfm?pageid=9716)، Fastpcr (http://www.biocenter.helsinki.fi/bi/Programs/fastpcr.htm)، Net primer (http://premierbiosoft.com/netprimer/index.html).
البته بسیاری از افراد با پیروی از قوانین ساده زیر، پرایمر موردنیازشان را طراحی میکنند. همان طور که قبلا نیز اشاره شد، یک پرایمر مناسب باید:
- طولی در حدود ۱۶ تا ۳۰ نوکلئوتید داشته باشد. همان طور که گفته شد، چنین طولی اختصاصیت مناسبی برای توالی هدفی منحصر به فرد فراهم میکند، حتی اگر نمونه DNA مورد استفاده به پیچیدگی DNA ژنومی انسان باشد.
- دارای مقادیر تقریبا برابری از هر نوکلئوتید باشد.
- حاوی توالیهای تکراری و یا نواحی حاوی تکرارهای پشتسرهم یک نوکلئوتید نباشد.
- از تکرار بازهای G و یا C به میزان ۳ بار و یا بیشتر جلوگیری شود؛ این کار منجر به جفت شدنهای نادرست در نواحی غنی از G-C میگردد.
- پرایمر نباید به علت مکمل بودن بازهای داخلی آن قادر به تشکیل ساختارهای ثانویه باشد.
- توالی انتهای ۳’ پرایمر نباید باعث باعث تشکیل جفتباز با خود و یا پرایمرهای دیگر شود؛ در غیر این صورت دایمرهای پرایمر تشکیل خواهند شد.